Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Dhanush Haspula and Version 2 by Camila Xu.
The hypothalamus and brainstem are critical components of the homeostatic system that regulates appetite and energy balance. These key brain regions comprise of distinct neuronal populations and nuclei which exerts tremendous control over several facets of energy balance. Importantly, several of these neuronal populations exhibit both overlapping and also contrasting metabolic roles, thereby enabling the CNS to fine tune metabolic functions under physiological conditions.
- obesity
- hypothalamus
- appetite
- glucose homeostasis
- weight-loss drugs
1. Hypothalamus
The hypothalamus is one of the most well-studied brain regions in metabolism. Apart from regulating a broad range of thermoregulatory, reproductive, and cardiovascular functions, it also exerts tremendous influence on several aspects of energy balance. The hypothalamus is composed of multiple nuclei located adjacent to the third ventricle. These nuclei comprise a distinct subpopulation of neurons, capable of altering energy intake and/or energy expenditure via anabolic or catabolic functions. The complex nexus of hypothalamic neuronal interconnections can integrate responses from peripheral signals (hormones, nutrients, and metabolites), to modulate appetite centrally, and to influence lipid and glucose metabolism, peripherally. Additionally, they also have reciprocal projections to and from extrahypothalamic nuclei located in the brainstem, midbrain, and forebrain which can also alter synaptic activity in the hypothalamic metabolic circuits. Consequently, via integration and coordination of responses from the brain and periphery, hypothalamic nuclei are key regulators of energy homeostasis.
1.1. Arcuate Nucleus (ARC)
The ARC is considered as one of the most important brain regions involved in the regulation of appetite and energy expenditure. Located near the median eminence, a region enriched in fenestrated capillaries, the ARC is accessible to circulating hormones, nutrients and metabolites, thus, serving as an ideal relay center to communicate circulating peripheral signals to the brain. The ARC comprises two distinct neuronal subpopulations that have opposing roles in energy homeostasis, the anabolic neuropeptideY/Agouti-related protein (NPY/AGRP) neurons and the catabolic, pro-opiomelanocortin (POMC) and cocaine-and amphetamine-regulated transcript (CART) or POMC/CART neurons (referred to henceforth as AGRP and POMC neurons). Both these neurons are first order neurons, which have glucose and nutrient sensing capabilities, in addition to receiving input from circulating hormones and satiety signals [1][2][3][4][8,9,10,11]. These counterregulatory neuronal populations are modulated by energy status. Food deprivation rapidly activates AGRP neurons and inhibits POMC neurons [5][6][7][12,13,14]. AGRP neurons release both NPY, which is an agonist for the Y1-5 receptors, and AGRP, an inverse agonist for melanocortin receptors [8][9][15,16]. Ablation of AGRP neurons results in a dramatic reduction in feeding, while acute activation results in a robust increase in food intake, weight gain, and altered autonomic outflow to several organs and tissues [10][11][12][13][17,18,19,20]. NPY was one of the first orexigenic neuropeptides to be identified, and subsequent functional studies revealed a potent, albeit fleeting, appetite-stimulating effect [14][21]. More recently, it has been revealed that NPY-mediated effects on feeding are mediated via the Y1 receptor, while its effects on energy expenditure are driven via the Y2 receptor [15][22]. Although both of these orexigenic neuropeptides, NPY and AGRP, have complimentary roles in triggering a hyperphagic response and reducing energy expenditure, the longer-lasting or sustained effect of these neurons on food intake is dependent on AGRP release, while the more rapid effect on food intake is dependent on NPY secretion [13][14][16][20,21,23]. Additionally, AGRP neurons also release GABA, which plays an integral role in AGRP-mediated effects on appetite and energy balance [14][17][21,24]. Furthermore, diet-induced obesity blunts AGRP responsiveness to circulating hormones [18][25]. In stark contrast to AGRP neurons, POMC neurons have a pronounced catabolic effect due to their ability to release the anorectic neuropeptide, α-melanocyte-stimulating hormone (α-MSH), a major satiety neuropeptide which is an agonist of melanocortin receptors [19][26]. Ablation of POMC neurons was reported to result in a mild obesity phenotype characterized by both reduced and increased food intake [20][21][27,28]. Interestingly, only chronic, but not acute chemogenetic activation of these neurons results in suppression of food intake, suggesting a role for POMC in maintaining long-term energy homeostasis [21][28]. POMC neurons have been reported to exhibit functional and spatial heterogeneity characterized by differences in both molecular architecture and anatomical projections to distinct brain regions, suggesting a more complex neural network involved in metabolic control [22][23][24][29,30,31]. POMC neuronal activity is also regulated by AGRP neurons. Anatomic and functional evidence indicates that GABA-releasing AGRP neurons are involved in inhibiting POMC neuronal activity and α-MSH release [25][26][27][32,33,34]. Apart from α-MSH, it is also to be noted that POMC neurons also release β-endorphin, which binds to the μ-opioid receptors. Both these POMC-derived neuropeptides have functionally antagonistic roles in the regulation of energy balance [28][35]. Both hypothalamic AGRP and POMC neurons are known to express the μ-opioid receptors (MOR). In the case of POMC neurons, the MORs function as autoinhibitory receptors that are activated by the release of β-endorphins [29][36]. Interestingly, while α-MSH is predominantly involved in suppressing appetite, β-endorphin were shown to play a major role in promoting a palatability-driven feeding response [30][31][37,38]. Naltrexone, a MOR antagonist which has been shown to suppress feeding on a short-term basis, has been shown to have stimulatory effects on POMC neurons in both rodents and humans [32][33][39,40]. More about their therapeutic utility will be covered in a later section.
Both AGRP and POMC neurons also express receptors for insulin (IR) and leptin (LepR). Leptin depolarizes and increases firing frequency of POMC neurons, while hyperpolarizing and inhibiting AGRP/NPY neuronal activity and neuropeptide release [34][35][36][37][38][41,42,43,44,45]. Mechanistic studies revealed that deletion of Rho-kinase 1, a protein kinase involved in cytoskeletal reorganization and neuropeptide release, in both AGRP and POMC neuronal populations resulted in leptin resistance and obesity [39][40][46,47]. Collectively, these data point to a crucial central mechanism by which leptin can induce a negative energy balance. Studies investigating the role of insulin signaling in both AGRP and POMC neurons on appetite regulation have yielded contradictory results. While some studies reported on little-to-no effect on appetite and body weight change with IR deletion in AGRP neurons, others have described a more nuanced role of AGRP-specific insulin signaling on regulating meal size [41][42][48,49]. A context-dependent appetite suppression role is reported for insulin signaling in the AGRP neurons, which is characterized by acute repression of feeding bouts without altering total calorie intake, and the suppression of highly palatable high-fat-diet food over standard chow [42][49]. In the case of POMC neurons, while the deletion of LepR results in mild obesity, knockout of IR in these neurons had no significant effect on body weight [43][44][50,51]. Furthermore, both AGRP and POMC neurons are modulated by postprandial signals, such as ghrelin, incretins, and amylin, to regulate food intake [45][46][47][48][49][52,53,54,55,56]. Apart from having integral roles in appetite and satiety regulation, these neuronal populations are also involved in maintaining glucose homeostasis as chemogenetic activation of AGRP and POMC neurons revealed distinct roles of G protein activation on food intake and glycemic control [11][13][21][50][18,20,28,57]. Additionally, both AGRP and POMC neurons can also regulate energy balance via the hypothalamic–pituitary–thyroid (HPT) axis. HPT axis is well-known to stimulate energy expenditure. Thyroid hormones play an important role in maintenance of homeothermia, and stimulation of the thyroid axis is known to increase energy expenditure via thermogenesis [51][58]. ICV administration of NPY has been shown to suppress circulating levels of thyroid hormones [52][59]. Interestingly, the melanocortin system has also been shown to regulate the HPT axis. Both in vivo and in vitro studies revealed that α-MSH can stimulate the HPT axis by increasing the levels of thyroid stimulating hormone (TSH), while AGRP on the other hand inhibits it [53][54][60,61]. For more information on the role of the melanocortin system in regulating the HPT axis, readers can refer to other reviews on this topic [55][62].
Another key aspect of the ARC neurons, especially POMC, is that they exhibit sexual dimorphism. Higher number of POMC neurons and increased neural activity were observed in female animals when compared to their male counterparts [56][63]. Disruption of key genes in POMC neurons in female mice resulted in the development of obesity [56][57][58][59][63,64,65,66]. More recently, POMC-specific alteration of certain highly expressed CNS genes, resulted in changes in glucoregulation and energy balance in female mice only [60][61][62][67,68,69].
ARC is highly susceptible to synaptic plasticity in response to the hormonal milieu. Both AGRP and POMC neurons have been described as exhibiting some level of synaptic rewiring under periods of food deprivation and overfeeding conditions [63][70]. Particularly, the melanocortin system has been reported to exhibit synaptic remodeling under both extreme metabolic changes, such as starvation and overfeeding, but also under physiological feeding states which results in modest metabolic changes [64][65][66][71,72,73]. Plasticity of the ARC has important implications in obesity, as diet-induced obesity has been demonstrated to suppress hypothalamic remodeling and neurogenesis resulting in reduced neuronal turnover [67][74]. It was also demonstrated to result in reactive gliosis in the ARC with altered synaptic architecture of the NPY and POMC neurons [68][75]. High fat diet (HFD)-induced neurogenesis is not restricted to the neuronal populations alone in the ARC. HFD activated neurogenesis in the median eminence however leads to energy storage, while prevention of it results in a reduction in weight gain [69][76]. Stimulation of neurogenesis in response to HFD is observed in female mice and not in males, suggesting a sexual dimorphic nature of hypothalamic neurogenesis [70][77].
1.2. Paraventricular Nucleus (PVH)
The PVH serves as an important convergence/termination point for orexigenic and anorexigenic projections arising from the ARC and other hypothalamic regions. Neurons present in this region express two different types of melanocortin receptors subtypes (MC3R and MC4R) that can be activated by the melanocortin peptide, α-MSH [71][72][78,79]. α-MSH and AGRP, released from the ARC projections, can modulate PVH neuronal activity by either activating or antagonizing the melanocortin receptors, respectively [9][72][73][74][16,79,80,81]. Thus, these neurons provide counterregulatory inputs to fine tune energy balance in response to changes in the levels of circulating signals. PVH neurons express single-minded 1 (Sim1), a transcriptional factor required for PVH development and the maintenance of energy homeostasis [75][76][82,83]. Sim1 neurons have pronounced effects on satiety and energy homeostasis as both sim1 heterozygous mice, and inducible Sim1-deficient mice, exhibit hyperphagia leading to obesity [76][77][83,84]. A major subset of Sim1 neurons in the PVH express MC4R [43][72][50,79]. Mutations in the MC4R gene are a leading cause of monogenic forms of obesity, and MC4R variants have been linked to increased obesity in certain populations [78][79][80][81][85,86,87,88]. The MC4R/Sim1 neurons, located in the PVH, together with the POMC neuronal projections, arising from the ARC, form the melanocortin pathway in the hypothalamus. Stimulation of MC4R neurons in the PVH results in pronounced satiation effects and thereby can induce a negative energy balance and confer protection against obesity [43][82][83][84][50,89,90,91]. Interestingly, short-term administration of MC4R agonists can also increase resting energy expenditure and shift substrate utilization towards increased fat oxidation in obese individuals suggesting additional mechanisms through which the melanocortin pathway and Sim1 neurons induce a negative energy balance [85][92]. Knockdown of MC4R results in potential disruption of synaptic plasticity and attenuation of long-term potentiation in the PVH [86][93]. Perturbation of MC4R signaling in the PVH alone, or in both PVH and DMV results in hyperphagic obesity with reduced energy expenditure and defects in insulin sensitivity [82][87][89,94]. It is to be noted that MC4R-expressing neurons are not just located in the hypothalamic nuclei, but also located in the brainstem, intermediolateral cell column of the spinal cord, and autonomic neurons where they not only exert prominent cardiovascular effects, but also regulate metabolic functions including thermogenesis, glucose homeostasis and energy expenditure [88][89][90][91][95,96,97,98]. Interestingly, PVH not only comprises MC4R neurons, but also contains other neuronal populations such as prodynorphin-expressing neurons, which lack MC4R. These neuronal populations have comparable effects to the PVH-MC4R expressing neurons on regulating satiety [92][99]. Several such anatomically distinct neuronal populations have been identified in the PVH as having appetite-regulatory roles, which further highlights the complexity of this nucleus [93][94][95][96][100,101,102,103]. For a more detailed review on the pathophysiological roles of MC4R neurons, readers can refer to excellent reviews on this topic [97][98][104,105].
1.3. Ventromedial Nucleus of the Hypothalamus (VMH)
Despite having an inauspicious history in metabolism research, the VMH is still appreciated as one of the principal satiety centers in the brain [99][106]. Early studies have highlighted an important role of VMH in suppressing appetite [100][101][107,108]. Apart from regulating food intake, VMH neurons have also been associated with improvements in several metabolic parameters and conferring protection against obesity [102][103][109,110]. A major subset of VMH neurons express steroidogenic factor 1 (SF1), often serving as a biomarker to distinguish VMH from other hypothalamic nuclei. Similar in function to the POMC neurons, activated SF1 neurons elicit pronounced anorexigenic effects with increased energy expenditure [104][105][111,112]. These neurons not only provide excitatory input directly onto the POMC neurons, but also project to the paraventricular thalamus to induce an aversive effect and suppress appetite [106][107][113,114]. Deletion of LepR from SF1 neurons also resulted in a similar degree of weight gain in mice when compared with LepR-specific KO in POMC neurons, suggesting important roles of leptin signaling in both sets of neuronal populations [43][108][50,115]. Moreover, SF1 neurons have distinct projections to other regions of the brain involved in negating insulin-induced hypoglycemia [109][116]. This will be covered in a later section. For a more detailed review on the role of SF1 neurons in metabolic disorders, the readers can refer to the review by Fosch et al. [110][117].
1.4. Dorsomedial Hypothalamus (DMH)
Another hypothalamic nucleus that affects feeding response is the DMH. DMH lesion in both young and older rats produced a hypophagic response with reduced body weight [111][118]. Interestingly, the DMH expresses NPY, which shows altered levels in various models of obesity [112][113][114][119,120,121]. Overexpression of NPY in the DMH results in an increase in food intake, weight gain, and an obese phenotype under high-fat-diet conditions, while knockdown of NPY ameliorated these effects in obese mice [115][122]. Inhibitory GABAergic neurons projecting to the PVH have been proposed as a key mechanism for eliciting a DMH-mediated orexigenic response [116][123]. Additionally, DMH neurons project to the ARC where they inhibit POMC neurons during fasting suggesting parallel neural circuits from DMH to regulate appetite [7][14]. The DMH may also be involved in the regulation of food intake by other hormones and peptides, as intra-DMH administration of the appetite-suppressing hormone, cholecystokinin (CCK), resulted in a suppression of food intake [117][118][124,125]. Interestingly, under refeeding conditions, excitatory glutamatergic projections are also activated by a subset of DMH glutamatergic neurons leading to reduced food intake [119][126]. A recently published study reported on DMH having bidirectional effects on food intake, which receive key leptin-responsive projections from the AGRP neurons [120][127]. Thus, it is likely that DMH projections could participate in the fine tuning of energy intake by activating distinct inhibitory and excitatory projections to other hypothalamic nuclei.
2. Brainstem
The brainstem exerts significant control over autonomous biological functions. The medulla is a key brainstem structure which has prominent cardioregulatory and metabolic functions, via specialized cardiovascular and satiety centers, respectively. The medullary cardiovascular centers have well-established roles in the homeostatic regulation of blood pressure via the baroreflex [121][128]. Although the brainstem is not as well-investigated as its counterpart, the hypothalamus, in metabolism, studies dating back to the 1970s highlighted the importance of the caudal brainstem in mediating satiety and glucoregulatory responses [122][123][129,130]. Importantly, specialized medullary regions serve as crucial integration points between the CNS and the digestive tract. They receive visceral afferent input from gastrointestinal sensory neurons, the latter conveying satiety signals in response to a meal. Additionally, the brainstem also comprises a circumventricular organ, area postrema (AP), which allows access to satiety signals. These signals in turn can modulate adjacent and supra-adjacent neuronal populations located in the brainstem. These proximally located neuronal populations in the caudal brainstem, in conjunction with the AP, are key structures in mediating postprandial satiety [124][125][131,132].
Dorsal Vagal Complex (DVC)
The caudal brainstem not only expresses receptors for circulating pressor peptides, but it can also be modulated by metabolic cues and thus exerts control over energy homeostasis [126][127][128][129][130][133,134,135,136,137]. The DVC located in the hindbrain is designated as the brainstem satiety center. The DVC comprises the AP, the nucleus of the solitary tract (NTS), and the dorsal motor nucleus of the vagus nerve (DMV). The NTS serves as the primary hub for ascending neural signals from the nodose ganglia, which contains cell bodies for several vagal afferents that densely innervate the gastrointestinal tract (GIT) [131][138]. The sensory vagal nerve terminals in the GIT are heterogenous in nature conveying both chemosensory, from nutrients and gut hormones, and mechanosensory signals to the brainstem [131][132][138,139]. Postprandial gut hormones and nutrients suppress food intake by transmitting information via the sensory vagal afferent terminals to the NTS, a crucial entry point in the brain for visceral information [133][134][140,141]. CCK, one of the first gut peptides to be identified to mediate satiety, elicits its actions by acting on the CCK-A receptors that are abundantly expressed on the vagal afferents and the cell bodies of the nodose ganglia [133][135][136][137][140,142,143,144]. Additionally other receptors involved in regulating satiety, such as the LepR, are also expressed on these cell bodies [138][145]. As a result, circulating signals such as leptin can also act along with CCK on the nodose ganglia, to synergistically suppress food intake [139][140][146,147]. Another class of gut hormones, the incretins, also exert prominent effects on satiety and glucose homeostasis. The incretins, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), are released in response to a meal, and they act on their corresponding receptors (GLP1R and GIPR) located in pancreatic islets where they promote insulin release. These receptors are also expressed in non-islet cells, where they exert prominent metabolic actions independent of direct effects on pancreatic insulin secretion [141][148]. In the GIT, the GLP1R is expressed on mechanosensitive vagal sensory neurons, and its activation results in pronounced inhibition of food intake, while its knockdown is associated with an increased meal size [142][143][149,150].
Interestingly, profiling of G-protein coupled receptors in the GIT revealed a lack of receptor expression for GIP and ghrelin on vagal afferents, potentially highlighting other CNS-dependent mechanisms to alter food intake [144][151]. Similar to the ARC, the AP lacks a well-defined blood–brain barrier, as a result is accessible to various satiety signals and circulating hormones. Since the NTS is located in close proximity to the fourth ventricle, it serves as a crucial node for integrating signals from the gut and circulation. Satiety signals, such as CCK, GLP-1, and their analogs, have been shown to inhibit appetite by acting on their corresponding receptors localized to the brainstem neurons in the satiety center [145][146][147][152,153,154]. The GLP1R is highly expressed in the NTS, and knockdown of preproglucagon, a precursor for GLP1, in the brainstem results in hyperphagia and increased adiposity, suggesting a crucial role for central GLP1R in mediating satiety [148][155]. Interestingly, GIPR agonism not only enhances the anorectic effect of GLP1R agonism, but recent studies suggest that it improves tolerability of PYY analogs by modulating the brainstem neural circuits and blocking its anorectic effect [149][150][151][156,157,158]. Thereby, understanding of incretins-mediated modulation of the brainstem neural circuitry has significant implications for the development of weight loss drugs with an improved side effect profile. The role of incretins in the hypothalamic and brainstem neural circuitry in the regulation of energy homeostasis will be discussed in a later section. Other pancreatic and gut-derived postprandial signals, such as amylin and PYY, also act on neuronal populations in the AP and NTS to promote satiety [152][153][154][155][156][159,160,161,162,163]. Additionally, leptin-mediated signaling in the NTS also activates the satiation neural circuitry to suppress food intake and regulate energy balance [157][158][159][164,165,166].
Projections from the NTS extend to other brain regions involved in appetite control and food aversion behaviors, where they suppress appetite by triggering either a positive or negative valence [160][161][167,168]. The latter may well be dependent on both the molecular architecture of the neural circuit, and the brain regions innervated by it. For instance, NTS projections to calcitonin gene-related protein (CGRP) expressing neurons located in higher brain regions, are strongly involved in mediating anorexia and reducing body weight [162][163][169,170]. However, they can exert opposing motivational valences, since projections to specific brain regions can generate both a positive valence (NTS to PVH projection) and a negative valence (NTS to PBN projection); the latter aversive response triggered by the activation of CGRP neurons in the PBN [164][165][166][171,172,173]. In stark contrast to the CCK neurons, calcitonin receptor expressing neurons from the NTS do not activate CGRP neurons, and hence produce a non-aversive suppression of food intake despite projecting to the PBN [166][173]. Other neuronal populations such as GLP-1 expressing neurons, which are primarily located in the caudal NTS, have projections to the VTA where they regulate intake of highly palatable food [167][168][174,175].
The NTS neurons also comprise a small, but metabolically relevant, population of POMC expressing neurons, accounting for about 10% of the total POMC neuronal population [169][170][176,177]. Interestingly, while they are activated by postprandial visceral afferents from the gut, they do not co-express several of the other neuropeptide markers observed in the NTS, suggesting a distinct hub of neurons involved in mediating satiety [171][172][178,179]. These neuronal populations are functionally similar to the POMC neurons in the ARC, but they exhibit different kinetics in terms of suppression of food intake. ARC-POMC neurons are involved in long-term suppression, while the NTS-POMC neurons mediate short-term feeding responses [21][28]. The latter neurons are potentially involved in a more rapid feeding suppression via circulating satiety signals. NTS-POMC neurons have been shown to be crucial for the acute appetite-suppressing effect of lorcaserin, indicative of their clinical relevance [173][180]. More recently, this effect of lorcaserin was also shown to be meditated via the GLP-1 neurons in the brainstem, in addition to the NTS-POMC neurons [174][181].
In addition to the regulation of food intake, the hindbrain circuitry also has important roles in glucose sensing and modulation of systemic glucose via vagal efferents [175][176][182,183]. Neuropeptide FF (NPFF), a key analgesic peptide which has been demonstrated to have a role in substrate utilization and regulation of energy balance, is strongly expressed in the caudal brainstem, mainly localized in the DVC [177][184]. More recently, a study reported on impairments in glucose homeostasis in mice deficient in NPFF, further highlighting the glucoregulatory role of the DVC [178][185]. The role of the various satiety signals in regulating glucose and lipid metabolism via brainstem circuits will be covered in more detail in later sections.
The sections so far highlight the pivotal roles of hypothalamic orexigenic and anorexigenic neuronal populations, along with the brainstem satiety center, in the regulation of energy intake and expenditure. A schematic summarizing this is shown in Figure 1. While the NTS integrates multiple metabolic cues to promote satiation, the ARC neuronal populations are able to exert both short- and long-term effects on energy homeostasis in response to energy demands.
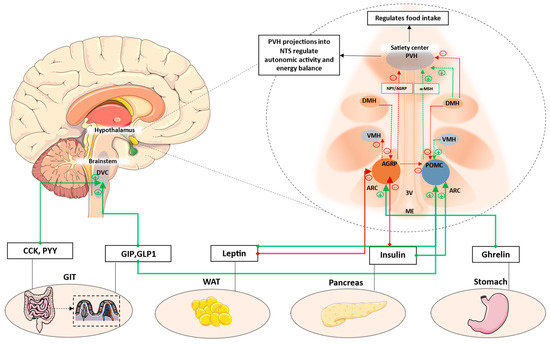
Figure 1. Key hypothalamic nuclei involved in the regulation of appetite and energy balance. ARC, comprising AGRP and POMC neurons, is located next to the median eminence. This region comprises permeable capillaries, thereby allowing access to circulating signals. These signals can modulate ARC neuronal populations, which then have extensive projections to PVH and other hypothalamic nuclei. PVH is the major hypothalamic satiety center. POMC neurons activate MC4R neurons in the PVH to decrease appetite, while AGRP neurons inhibit PVH-MC4R neurons to increase appetite. Additionally, AGRP neurons also inhibit POMC neurons via stimulation of inhibitory GABAergic input to POMC neurons. Anorexigenic signals such as leptin and GLP1 increase satiety by acting on POMC neurons, whereas orexigenic signals such as ghrelin can increase appetite by acting on AGRP neurons. Other hypothalamic neuronal populations have extensive projections to and from adjacent nuclei. While DMH has predominantly inhibitory projections to PVH and POMC, it also has been shown to also have activate inhibitory GABAergic neurons projecting to the AGRP neurons in the ARC. VMH mainly has excitatory projections to the POMC neurons, while AGRP neurons has inhibitory projections to VMH. Additionally, postprandial satiety signals from the enteroendocrine cells of the GIT can also act on DVC located in the brainstem to suppress appetite. AGRP: Agouti-related protein; POMC: Pro-opiomelanocortin; ARC: Arcuate nucleus; ME: median eminence; VMH: Ventromedial nucleus of the hypothalamus; DMH: Dorsomedial hypothalamus; PVH: Paraventricular nucleus; 3V: Third ventricle; DVC: Dorsal vagal complex; CCK: cholecystokinin; GIP: Glucose-dependent insulinotropic polypeptide; GLP1: Glucagon-like peptide-1; WAT: White adipose tissue; GIT: Gastrointestinal tract. Green dotted lines/arrows represent activation. Red dotted lines/arrows represent inhibition.