Cystic fibrosis (CF) is the rare genetic disease caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR). These molecules, known as CFTR modulators, have led to unprecedented improvements in the lung function and quality of life of most CF patients. However, the efficacy of these drugs is still suboptimal, and the clinical response is highly variable even among individuals bearing the same mutation. Furthermore, not all patients carrying rare CFTR mutations are eligible for CFTR modulator therapies, indicating the need for alternative and/or add-on therapeutic approaches. Because the second messenger 3′,5′-cyclic adenosine monophosphate (cAMP) represents the primary trigger for CFTR activation and a major regulator of different steps of the life cycle of the channel, there is growing interest in devising ways to fine-tune the cAMP signaling pathway for therapeutic purposes.
- cystic fibrosis
- cystic fibrosis transmembrane conductance regulator
- 3′,5′-cyclic adenosine monophosphate
1. Introduction
2. cAMP Is Recognized as the Master Regulator of CFTR Activity
cAMP elevation in the subcortical compartment leads to the activation of PKA and PKA-mediated phosphorylation of the CFTR is required to increase the channel open probability and to allow efflux of Cl– anions [10]. In epithelial cells, the cAMP pool responsible for CFTR function is primarily produced upon the activation of a specific GPCR, namely the β2AR channel (Figure 1). This protein co-localizes with the CFTR at the apical membrane of polarized lung epithelial cells (Calu-3), and it has also been found to co-immunoprecipitate with the channel itself [26,27][12][13]. The physical protein-protein interaction (PPI) between the anion channel and the β2AR is coupled with CFTR activation since βAR agonists, such as isoproterenol (β1AR and β2AR agonist) and albuterol (β2AR-selective agonist), can induce a concentration-dependent increase in short-circuit current (ISC), which is sensitive to CFTR-selective inhibition in polarized epithelial cells [26][12]. It is also notable that the β2AR-dependent CFTR opening can be stimulated only upon the interaction between the two proteins, since the removal of the PDZ-binding motif of CFTR, which abolishes the physical interaction between the channel and the receptor, specifically reduces Cl− efflux after β2AR stimulation in vitro [26][12]. The relevance of β2ARs to CFTR activation is corroborated by the observation that β2AR agonists induce the swelling of intestinal organoids, a gold-standard model in CF research, indicating that these molecules can potently activate the wild-type (wt) CFTR [28][14]. Again, in intestinal epithelial cells, it has been shown that AC6 plays a central role in regulating CFTR function upon GPCR activation (Figure 1). AC6, the most abundant AC isoform in the gut, can be detected as a CFTR interactor in the intestinal mucosa of mice and in colon epithelial cells [29][15]. In further support of the key role of AC6 in producing the cAMP pool responsible for CFTR activity in the gut, Thomas and colleagues showed that the AC activator forskolin failed to induce the swelling of intestinal organoids derived from AC6 knockout mice [29][15]. Another well-characterized interactor with the CFTR protein is PKA, one of the main effectors of the second messenger cAMP. PKA phosphorylates the CFTR protein on several serines mainly located within the regulatory R domain of the channel (Figure 1), an unstructured polypeptide sequence with a predominant inhibitory function. In its unphosphorylated state, the R domain intercalates between the two nucleotide-binding domains (NBDs), preventing their dimerization and CFTR opening [30][16], whereas phosphorylation of residues S422, S660, S795, and S813, can release the channel from its closed conformation, increasing its open probability up to 100-fold [31][17]. Unlike the aforementioned serines, whose post-translational modification is considered as being activating, some residues such as S737 and S768 have been proposed to be either activating or inhibitory, depending on contextual modifications of other phosphorylation sites [32][18]. In this regard, the phosphorylation of different serine residues can be interdependent, as seen in S795 and S813. The latter can be phosphorylated only when S795 has already been post-translationally modified, showcasing S813 phosphorylation as a limiting step in CFTR activation [33][19].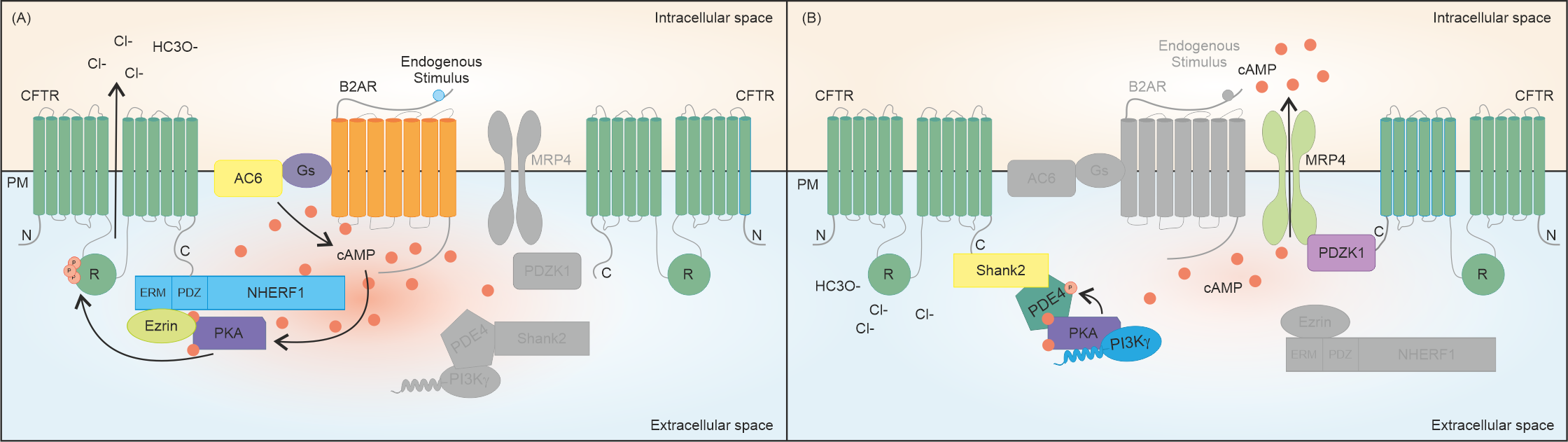
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211.
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castanos, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124.
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948.
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819.
- Orenti, A.; Zolin, A.; Jung, A.; van Rens, J.; Adamoli, A.; Prasad, V.; Fox, A.; Krasnyk, M.; Lorca Mayor, S.; Naehrlich, L.; et al. ECFSPR Annual Report 2021. 2023. Available online: https://www.ecfs.eu/sites/default/files/Annual%20Report_2021_09Jun2023.pdf (accessed on 16 June 2023).
- Fajac, I.; Sermet, I. Therapeutic approaches for patients with cystic fibrosis not eligible for current CFTR modulators. Cells 2021, 10, 2793.
- Graeber, S.Y.; Vitzthum, C.; Pallenberg, S.T.; Naehrlich, L.; Stahl, M.; Rohrbach, A.; Drescher, M.; Minso, R.; Ringshausen, F.C.; Rueckes-Nilges, C.; et al. Effects of elexacaftor/tezacaftor/ivacaftor therapy on CFTR function in patients with cystic fibrosis and one or two F508del alleles. Am. J. Respir. Crit. Care Med. 2022, 205, 540–549.
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983.
- Farinha, C.M.; Gentzsch, M. Revisiting CFTR interactions: Old partners and new players. Int. J. Mol. Sci. 2021, 22, 13196.
- Chin, S.; Hung, M.; Bear, C.E. Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants. Cell Mol. Life Sci. 2017, 74, 57–66.
- Murabito, A.; Cnudde, S.; Hirsch, E.; Ghigo, A. Potential therapeutic applications of AKAP disrupting peptides. Clin. Sci. 2020, 134, 3259–3282.
- Naren, A.P.; Cobb, B.; Li, C.; Roy, K.; Nelson, D.; Heda, G.D.; Liao, J.; Kirk, K.L.; Sorscher, E.J.; Hanrahan, J.; et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. USA 2003, 100, 342–346.
- Trotta, T.; Guerra, L.; Piro, D.; d’Apolito, M.; Piccoli, C.; Porro, C.; Giardino, I.; Lepore, S.; Castellani, S.; Di Gioia, S.; et al. Stimulation of beta2-adrenergic receptor increases CFTR function and decreases ATP levels in murine hematopoietic stem/progenitor cells. J. Cyst. Fibros. 2015, 14, 26–33.
- Vijftigschild, L.A.; Berkers, G.; Dekkers, J.F.; Zomer-van Ommen, D.D.; Matthes, E.; Kruisselbrink, E.; Vonk, A.; Hensen, C.E.; Heida-Michel, S.; Geerdink, M.; et al. beta2-Adrenergic receptor agonists activate CFTR in intestinal organoids and subjects with cystic fibrosis. Eur. Respir. J. 2016, 48, 768–779.
- Namkung, W.; Finkbeiner, W.E.; Verkman, A.S. CFTR-adenylyl cyclase I association responsible for UTP activation of CFTR in well-differentiated primary human bronchial cell cultures. Mol. Biol. Cell 2010, 21, 2639–2648.
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular structure of the human CFTR ion channel. Cell 2017, 169, 85–95.e88.
- Mihalyi, C.; Iordanov, I.; Torocsik, B.; Csanady, L. Simple binding of protein kinase A prior to phosphorylation allows CFTR anion channels to be opened by nucleotides. Proc. Natl. Acad. Sci. USA 2020, 117, 21740–21746.
- Della Sala, A.; Prono, G.; Hirsch, E.; Ghigo, A. Role of protein kinase A-mediated phosphorylation in CFTR channel activity regulation. Front. Physiol. 2021, 12, 690247.
- Infield, D.T.; Schene, M.E.; Fazan, F.S.; Galles, G.D.; Galpin, J.D.; Ahern, C.A. Real-time observation of functional specialization among phosphorylation sites in CFTR. J. Gen. Physiol. 2023, 155, e202213216.
- Sun, F.; Hug, M.J.; Bradbury, N.A.; Frizzell, R.A. Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J. Biol. Chem. 2000, 275, 14360–14366.
- Li, J.; Dai, Z.; Jana, D.; Callaway, D.J.; Bu, Z. Ezrin controls the macromolecular complexes formed between an adapter protein Na+/H+ exchanger regulatory factor and the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2005, 280, 37634–37643.
- Guerra, L.; Fanelli, T.; Favia, M.; Riccardi, S.M.; Busco, G.; Cardone, R.A.; Carrabino, S.; Weinman, E.J.; Reshkin, S.J.; Conese, M.; et al. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 2005, 280, 40925–40933.
- Singh, A.K.; Riederer, B.; Krabbenhoft, A.; Rausch, B.; Bonhagen, J.; Lehmann, U.; de Jonge, H.R.; Donowitz, M.; Yun, C.; Weinman, E.J.; et al. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J. Clin. Investig. 2009, 119, 540–550.
- Lee, J.H.; Richter, W.; Namkung, W.; Kim, K.H.; Kim, E.; Conti, M.; Lee, M.G. Dynamic regulation of cystic fibrosis transmembrane conductance regulator by competitive interactions of molecular adaptors. J. Biol. Chem. 2007, 282, 10414–10422.
- Kim, J.Y.; Han, W.; Namkung, W.; Lee, J.H.; Kim, K.H.; Shin, H.; Kim, E.; Lee, M.G. Inhibitory regulation of cystic fibrosis transmembrane conductance regulator anion-transporting activities by Shank2. J. Biol. Chem. 2004, 279, 10389–10396.
- Blanchard, E.; Zlock, L.; Lao, A.; Mika, D.; Namkung, W.; Xie, M.; Scheitrum, C.; Gruenert, D.C.; Verkman, A.S.; Finkbeiner, W.E.; et al. Anchored PDE4 regulates chloride conductance in wild-type and DeltaF508-CFTR human airway epithelia. FASEB J. 2014, 28, 791–801.
- Ghigo, A.; Murabito, A.; Sala, V.; Pisano, A.R.; Bertolini, S.; Gianotti, A.; Caci, E.; Montresor, A.; Premchandar, A.; Pirozzi, F.; et al. A PI3Kgamma mimetic peptide triggers CFTR gating, bronchodilation, and reduced inflammation in obstructive airway diseases. Sci. Transl. Med. 2022, 14, eabl6328.
- Turner, M.J.; Sato, Y.; Thomas, D.Y.; Abbott-Banner, K.; Hanrahan, J.W. Phosphodiesterase 8A regulates CFTR activity in airway epithelial cells. Cell Physiol. Biochem. 2021, 55, 784–804.
- Nguyen, J.P.; Kim, Y.; Cao, Q.; Hirota, J.A. Interactions between ABCC4/MRP4 and ABCC7/CFTR in human airway epithelial cells in lung health and disease. Int. J. Biochem. Cell Biol. 2021, 133, 105936.
- Li, C.; Krishnamurthy, P.C.; Penmatsa, H.; Marrs, K.L.; Wang, X.Q.; Zaccolo, M.; Jalink, K.; Li, M.; Nelson, D.J.; Schuetz, J.D.; et al. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 2007, 131, 940–951.