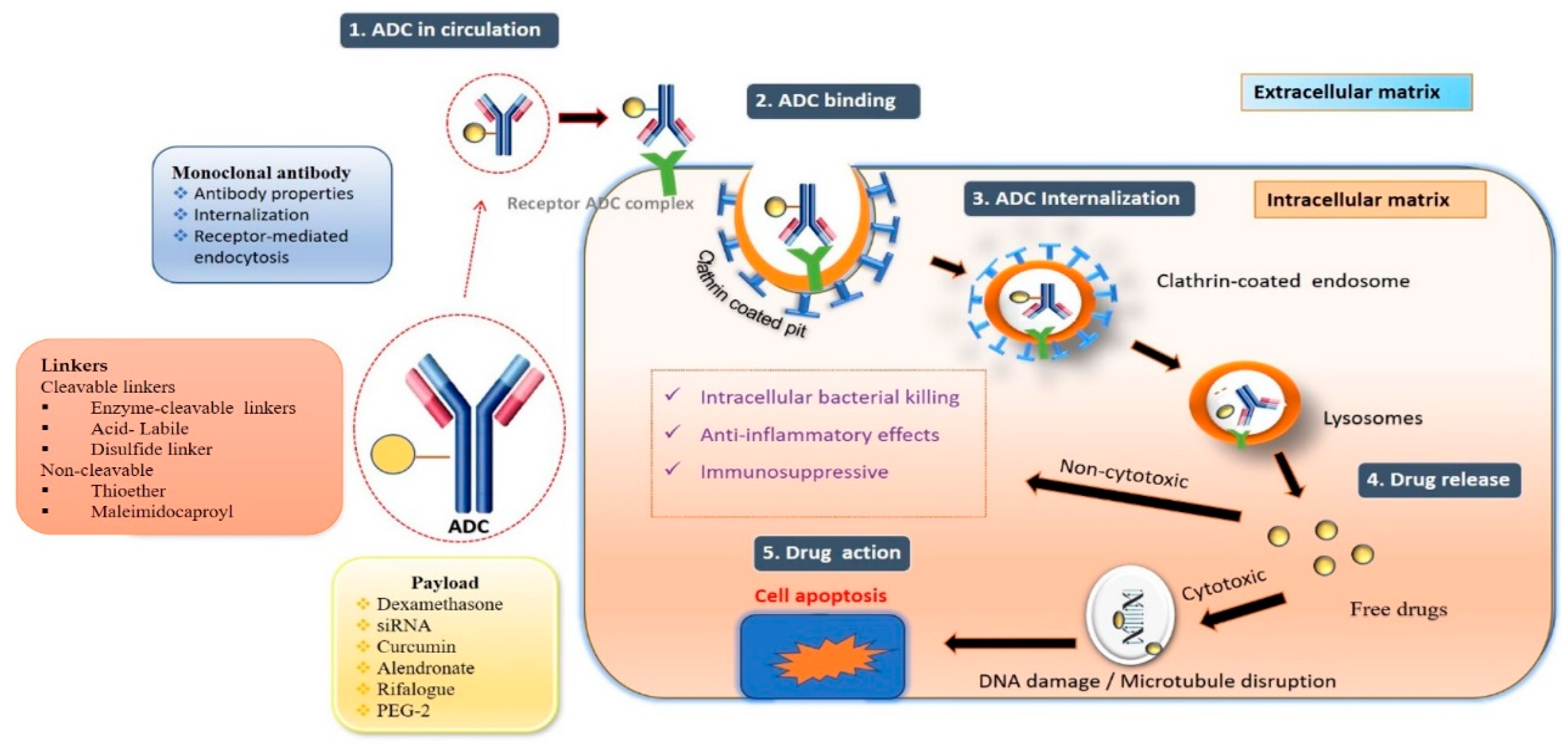
The substantial increase in ADCs has been seen in non-oncological applications, where the payload can change specific biological processes instead of directly killing the cells. For example, antibiotics as a payload can be used to treat bacterial infections, and glucocorticoids can be used to suppress the immune system.
2.2. Antibody-Mediated siRNA Conjugates
Small interfering RNA (siRNA)-based therapy has gained much curiosity from researchers due to its potency. However, concerns like adverse effects due to off-target exposure limit their application. Noncovalent hybridization or the direct oligonucleotide conjugation of siRNA to the antibodies or their fragments (Fab, scFv) generates the antibody–siRNA conjugates (ARCs). The antibody–siRNA conjugates (ARCs) show specific transport into tissues, cells infected with viruses, or cancer cells, both in vitro as well as in vivo. Cell surface receptors bind ARCs carrying Fab and siRNA, which are then taken up by endosomes and transported into the cell, where the siRNA is released and escapes into the cytoplasm. After the siRNA has been loaded into RISC (RNA-induced silencing complex), specific mRNA is degraded. This concept is utilized when nucleic acid is used as a payload
[7][47].
ADCs, due to their site-specific delivery, attracted the researchers to combine the antibodies with siRNA for an efficient therapeutic potential. In their recent article, Cao W et al.
[8][48] discussed the different approaches for conjugating siRNA with antibodies. They explained various conjugation mechanisms and different linkers used to conjugate the siRNA with antibodies. The authors also mentioned a few examples of ADCs and their application in multiple diseases.
2.3. Antibody–Antibiotic Conjugate
Antibiotics are often regarded as the hallmark of contemporary medicine. For decades, they have aided in preventing infectious disease epidemics and allowed for the safe practice of invasive surgery. The emergence of multi-drug resistance, along with a decline in the discovery and development of new antibiotics, has resulted in a global crisis with potentially disastrous repercussions. The application of ADCs in the treatment of challenging diseases such as cancer and autoimmune disorders has been very successful. A few researchers experimented with a similar concept and developed antibody–antibiotic conjugates (AACs), which combine the benefits of antibodies and antibiotics into a hybrid structure to address the limitations associated with antibiotic resistance
[9][10][51,52]. MRSA (Methicillin-resistant
S. aureus), resistant to all beta-lactam antibiotics, has emerged and expanded rapidly over the last few decades, making
S. aureus infections more challenging to treat
[11][53]. Lehar et al.
[12][29] demonstrated that intracellular MRSA is difficult to remove with standard antibiotics, including vancomycin, linezolid, and daptomycin, used against invasive MRSA infections.
S. aureus can invade phagocytic cells from there and also invades non-phagocytic cells, and this intracellular
S. aureus is responsible for many ailments. It is necessary to clear intracellular MRSA to improve clinical success.
The capacity of antibody–antibiotic conjugates to deliver antibiotics with undesirable pharmacokinetics or toxicity profiles was proven by Kajihara et al.
[13][57]. One of the difficulties in treating infectious diseases is the multidrug resistance (MDR) of bacteria, which raises mortality.
Pseudomonas aeruginosa is a gram-negative bacterium responsible for some pulmonary and bloodstream-related infections. The development of resistance to these bacteria increased mortality and posed a critical problem. The MDR
P. aeruginosa infection was recently designated as a “critical threat” pathogen by the World Health Organization. The authors selected mAb 26F8 as mAb with higher specificity and binding towards
P. aeruginosa after the preliminary analysis. Later, AAC was generated by conjugating the antibiotic (G2637) to the mAb modified with six unpaired cysteines. In the presence of lysosomal cathepsins, the linker cyclobutane-1,1-dicarboxamide citrulline breaks down to release the attached antibiotic. LC-MS/MS analysis showed a DAR of 6 for the generated AAC. The cleavage of the linker and release of antibiotic was analyzed after incubating the AAC with
P. aeruginosa in the presence and absence of cathepsin B. The presence of cathepsin B improved the level of inhibition compared to samples lacking cathepsin B, demonstrating that antibiotic release is a prerequisite for the AAC to function. This was further confirmed by quantification of the free antibiotic after adding the incubated AAC with
P. aeruginosa PA14 WT to RAW264.7 macrophages. Here also, AAC containing cleavable linkers showed higher intracellular antibiotic concentrations in the macrophages over the AAC with non-cleavable linkers. The use of AAC indicated that the concentration of antibiotic required will be two-fold less compared to free antibiotics, indicating the efficiency of AAC. The authors came to the conclusion that using AAC allowed for larger intracellular antibiotic concentrations, which resulted in greater antibiotic efficacy as compared to using the free medication.
2.4. Glomerular Nephritis
Current treatments must improve specificity and reduce systemic toxicity to slow the progression of kidney disease. Kvirkvelia et al.
[14][58] hypothesized that a human monoclonal antibody (F1.1) against the glomerular-localized noncollagenous-1 domain (NC1) of 3(IV) collagen may play the role of a carrier for precise drug administration. 3(IV)NC1 is a perfect target for the administration of disease-modifying medicines because of its augmented exposure during glomerular disorders and its subdued epitope expression in other organs.
The purified human monoclonal antibody was connected with the PGE2 or Dex through an amide bond formed between the carboxylic group of Dex or PGE2 and the mAb amide group using EDC (1-ethyl-3- [3-dimethylamino -propyl] carbodiimide hydrochloride) zero-length cross-linker. PGE2 and Dex were chosen because of their activity in nephrotoxic nephritis and anti-inflammatory properties, respectively. Using an ELISA kit for PGE2, they determined the amount of PGE2 bound to the antibody. Conjugate functionality was evaluated by measuring their ability to bind to podocytes using flow cytometry. The observation of low binding confirmed the antigenic specificity of the conjugates in additional cell lines (such as mouse mesangial cells and hepatocyte AML-12 cells). It inhibited the podocyte association by recombinant 3(IV).
2.5. Rheumatoid Arthritis (RA)
Synovial membrane inflammation and bone degradation are the characteristics of rheumatoid arthritis, a severe chronic inflammatory disorder. Autoimmunity and tissue damage in RA are brought by excess pro-inflammatory cytokines in the synovial fluid and blood
[15][16][59,60]. The pathophysiology of rheumatoid arthritis is so complex that its specific mechanism is not yet fully known. However, IL-6 signaling is a significant cause of inflammation and RA symptoms. Cells such as macrophages, osteoclasts, B cells, and T cells have IL-6 and respond to IL-6 signaling, contributing to the immune response in RA. The effectiveness of cytotoxic drugs used in RA therapy is limited by their side effects associated with non-specific targeting. Hence, a formulation strategy is needed to act specifically at the target site. Among the well-known targeted drug delivery strategies, ADCs have recently received much attention for their ability to dispense conjugated drugs to specific cells with negligible adverse drug reactions
[17][61].
2.6. Immuno-Suppression
Dasatinib was clinically used for treating BCR-ABL-dependent chronic myelogenous leukemia. It also has potential immunosuppressive activity due to the inhibition of Src-family kinases such as Lck and Fyn. However, due to a lack of selectivity, dasatinib has severe side effects that limit its clinical application as an immunosuppressing agent. Wang et al.
[18][21] developed an antibody–drug conjugate for the site-specific delivery of dasatinib to T lymphocytes for effective immunosuppression. The authors initially synthesized an antibody specifically targeting T cells and conjugated the antibody to dasatinib in the next step. Finally, the antibody–dasatinib conjugate was evaluated in vitro for immunosuppression activity. Initial screening was performed on several antibodies that could bind to T cells, and CD184 (CXCR4) was selected based on its high expression, better internalization, and fewer side effects. A CXCR4 antagonist was joined with a bovine antibody (BLV1H12) scaffold and further grafted into the CDR3H of trastuzumab to produce fewer immunogenic-humanized antibodies (HLCX). HEK 293F cells were used for the expression of HLCX. Further protein G chromatography was used to purify the antibody, and its mass was confirmed by electrospray ionization mass spectrometry.
A shift in the peak in flow cytometry analysis confirmed the binding of HLCX to CXCR4. The antibody was incubated with cell lines greatly expressing CXCR4 (Jurkat T cells) and cell lines lacking CXCR4 (MDA-MB435 cells). The samples incubated with Jurkat cells clearly showed a shift in the peak (96.2%) due to binding, whereas samples incubated with MDA-MB435 cells did not show any marked shift in the peak due to the absence of binding. This was further confirmed by analyzing the samples incubated with different cell lines with varying levels of CXCR4 expression. The internalization ability of the HLCX into T cells was confirmed by confocal microscopy. Alexa Fluor 488 (AF488) dye was conjugated to the HLCX and an unconjugated antibody and incubated with human T lymphocytes. Green spots were observed in the cytoplasm of T cells within 30 min after mixing at 37 °C, indicative of HLCX-AF488 internalization. This was not observed at 4 °C and with unconjugated HLCX, confirming the site-specific delivery of HLCX. Two ADCs, namely, non-cleavable (HLCX-dasatinib) and cleavable linkers (HLCX-SS-dasatinib), were synthesized in a two-step coupling process after modifying the dasatinib and antibody as both do not have interacting groups directly. Synthesized ADCs were >90% pure, and the drug-to-antibody ratio was 3, as confirmed by SDS phase analysis. The molecular weights were in the anticipated range, suggesting the structural integrity of the synthesized ADCs.
2.7. Atherosclerosis
Atherosclerosis is a condition developed due to the deposition of cholesterol in arteries, responsible for about 35% of deaths in the United States. The process involves the development of oxidized low-density lipoproteins (oxLDL) and a consequent inflammatory response and accumulation of macrophages. The athero-protective effect of macrophages is exerted by transferring cholesterol to the liver for bile secretion through reverse cholesterol transport (RCT). The RCT is triggered by liver X receptors (LXR-α and LXR-β) and others. LXR activation blocks macrophage proliferation and foam cell formation by suppressing pro-inflammatory cytokines. LXR agonists (GW3965 and T0901317) were developed and tested for their activity against atherosclerosis. However, these derivatives also showed increased triglycerides in the liver due to an overexpression of LXR-α. The site-specific delivery of LXR agonists to the macrophages may reduce this risk and improve the clinical applicability of LXR agonists. The ADC-based delivery of LXR agonists to macrophages was described by Lim et al.
[19][22]. The authors synthesized a new compound similar to the above-mentioned LXR agonists that can retain its antigenic nature after linking to an antibody with the substitution of aminoethyl sulfonamide. The protease cleavable linker was found to be suitable after screening the various linkers with disulfides, acid-labile hydrazones, and protease cleavable linkers. Using the chemistry approaches, they synthesized a stable aminooxy-CatB-LXR agonist and confirmed its cleavable potential in the presence of purified CatB enzyme using liquid chromatography and mass spectrometry (LC-MS). The authors selected CD11a as a particular antigen expressed on leukocytes, monocytes, and macrophages but not on the liver. After cloning the humanized antibody Efalizumab and transfecting the Chinese Hamster Ovary (CHO) suspension cells, the required antibody for conjugation, anti-CD11a IgG, was obtained. This was purified, and mass analysis was performed by SDS-PAGE analysis and electrospray-ionization mass spectrometry, respectively. In further experiments, two more antibodies, anti-Her2 IgGX and anti-Her2 FabX, were also expressed as negative controls. Finally, the linker attached to the LXR agonist was conjugated with anti-CD11a IgGX via covalent bonds.
The binding of the conjugate was compared with that of the negative control after incubation with human THP-1 monocyte/macrophage cells and human HepG2 hepatoma cells. Further Fc blockers were also used to confirm that the binding is CD11a-mediated. The flow cytometry analysis indicated anti-CD11a IgGX-AF488 binds to the monophages and macrophages as characterized by distinct peak shifts in flow cytometry analysis. It does not bind to the Hepatoma cells, as indicated by the absence of peak shift. Similarly, the binding of anti-CD11a IgGX-AF488 to monophages exists in the presence of Fc blockers, indicating the binding is CD11a-mediated and not Fc-controlled. Negative control antibody conjugates showed binding in the absence of Fc blockers, and the presence of blockers did not confirm the binding to the cells as they are Fc-dependent. The internalization of the conjugate was established from the identification of the dye (Alexa Fluor 488)-attached conjugate in the cytoplasm of the THP-1 cells after incubation, as observed under confocal microscopy.
The LXR Transactivation assay was used to examine the LXR agonistic activity of synthesized ADC and compared with that of positive control and T0901317 (LXR agonist) and aminooxy-CatB-LXR agonist. All three compounds were incubated with THP-1 cells and HEpG2 cells in the presence and absence of 10% human AB serum to block Fcγ receptors. Then, the cells were treated with ONE-Glo, and the luminescence was recorded on an Envision plate reader. The synthesized conjugate (anti-CD11a IgGX-AF488) demonstrated a strong agonistic action in THP-1 cells without any activity in HEPG2 cells. The positive control showed equivalent activity in both cell lines, and the aminooxy-CatB-LXR agonist showed no agonistic activity. This indicates that anti-CD11a IgGX-AF488 has a specific binding ability towards macrophages with three-fold higher potency.
2.8. Systemic Sclerosis
Systemic sclerosis (SSc) is an autoimmune disorder characterized by immune system dysregulation that causes fibrosis in the skin and viscera. There is no treatment for this disease at present, but treating the patients during the early stages to reverse the inflammation may provide relief. Recent studies revealed that there was a significant enhancement in the inflammatory markers, such as CD28-CD80/86, CD19, CCL24, CD20, CD30, tumor necrosis factor, transforming growth factor β, B-cell activating factor, lysophosphatidic acid receptor 1, soluble guanylate cyclase, Janus kinases, and interleukin 6 in the serum of SSc patients
[20][62]. The enhanced CD30 can be targeted using Brentuximab (αCD30 antibody). Based on the concept, three clinical trials were initiated (
clinicaltrials.gov accessed on 19 June 2023, identifierNCT03222492, NCT03198689, NCT05149768). The NCT05149768 trial is a dose-escalation safety study of brentuximab vedotin, a drug–antibody conjugate approved for lymphoma treatment and targeted to the protein CD30 molecule expressed in activated immune cells. There is evidence that CD30 plays a role in SSc. This study is the initial step in evaluating the safety and tolerability of brentuximab vedotin in SSc patients
[21][63]. NCT03198689 is designed to evaluate the feasibility, safety, and preliminary efficacy of Brentuximab vedotin (Adcetris)
[22][64]. The NCT05149768 trial is intended as a 48-week extension of the current therapy to make the treatment available for SSc patients who improved considerably with Brentuximab vedotin (24 weeks treatment) but relapsed after treatment termination
[23][65]. All three trails are in phase II and currently active. According to clinical trial data, the first two studies are expected to be completed by 2023 and the third by 2024.
A clinical trial was proposed on
clinicaltrials.gov, accessed on 19 June 2023 (identifier NCT01616680) for using Adcetris in Steroid-Resistant Acute Graft-Versus-Host Disease but it was withdrawn before recruiting the subjects
[24][66].