Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by GuangJun Zhang.
Developmental patterning is essential for regulating cellular events such as axial patterning, segmentation, tissue formation, and organ size determination during embryogenesis. Understanding the patterning mechanisms remains a central challenge and fundamental interest in developmental biology. Ion-channel-regulated bioelectric signals have emerged as a player of the patterning mechanism, which may interact with morphogens.
- zebrafish
- embryonic development
- long fin
- short fin
1. Introduction
Embryonic development is a self-autonomous and robust process in which a new body develops from a fertilized egg. This developmental process requires coordinated and complex cellular events such as proliferation, differentiation, and movement. The related patterning mechanisms are essential and instructive elements that eventually guide the body shape and organ sizes [1,2,3][1][2][3]. The morphogen gradient and transcription network are the mainstay theories and have been verified in many organ systems of various organisms [4,5,6][4][5][6]. Recent and past evidence revealed that ion-channel-related bioelectricity is a new component of the regulating mechanism for developmental patterning, regeneration, and cancers [7,8][7][8].
Bioelectricity is defined as endogenous electrical signaling across cell membranes and is mediated by the dynamic distribution of charged molecules [7,8,9,10,11,12,13][7][8][9][10][11][12][13]. This is represented by a difference in the net charge of cations and anions inside versus outside a cell. Many components are involved in electrical potential formation [12,14][12][14]. In essence, the semipermeable lipid-based plasma membrane acts as an electrical insulator, but also as a capacitor that can accumulate charge, while specialized passages (ion channels, pumps, connexins/gap junctions, and solute carriers) regulate ion flow from one side to the other, altering the voltage of the cell (Figure 1A). All cell types form ionic gradients across their cell membranes because channels exist throughout all living organisms in all domains of life, including plants, fungi, and bacteria [14,15,16,17,18,19,20,21,22,23,24][14][15][16][17][18][19][20][21][22][23][24]. Thus, ion regulation and the resulting bioelectricity are considered essential properties of living cells across evolution, and their innate properties can be used for cellular communication [25,26][25][26]. Therefore, understanding additional aspects of bioelectricity in cells and organisms is fundamental for modern physiology and ontology.
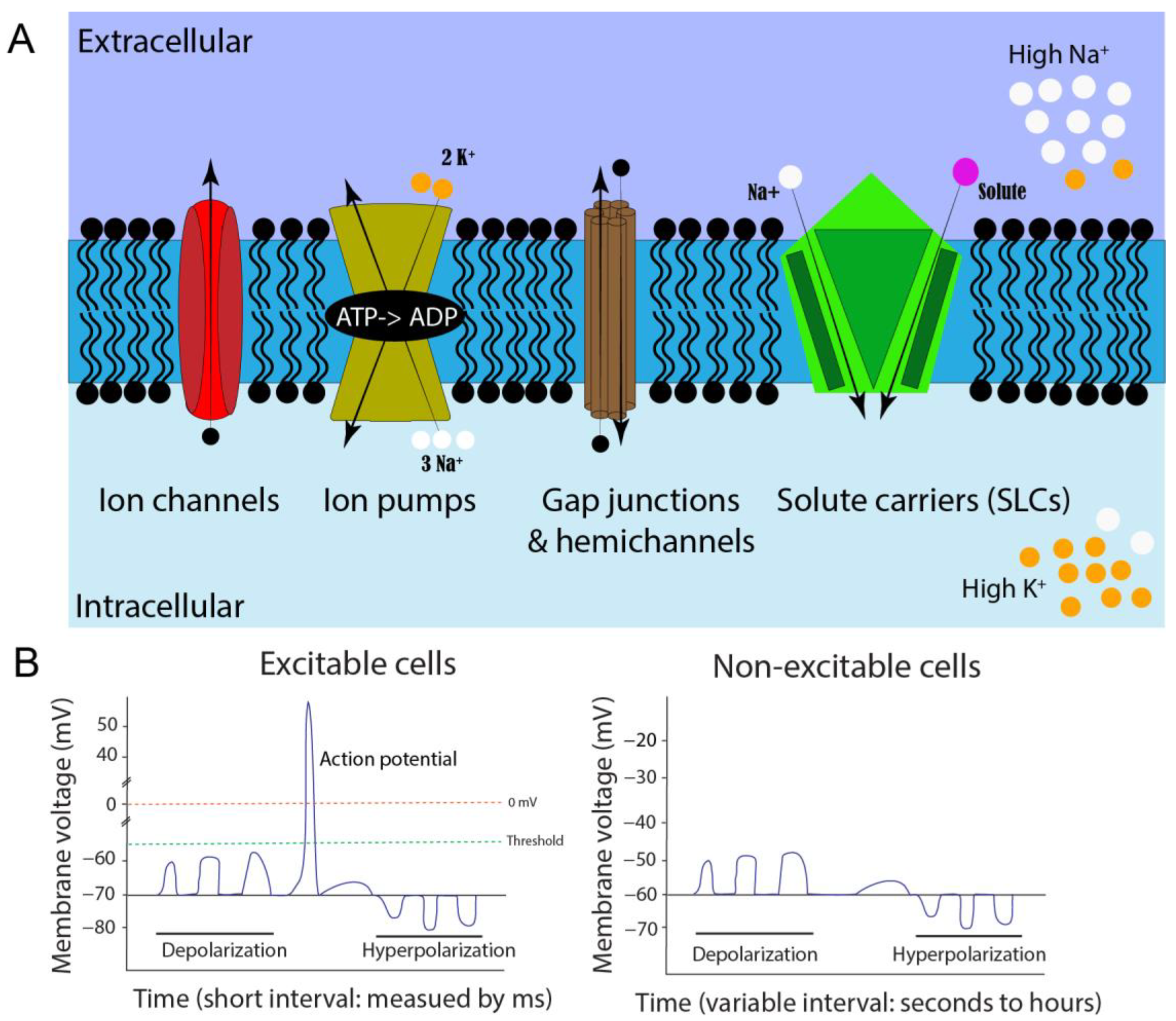
Figure 1. Cell membrane potential formation and comparison of neuromuscular excitable cells and non-excitable somatic cells. (A). Illustration of resting membrane potential, ion regulators, and ionic concentrations when the cell is in a non-excitable state. Different shapes represent various ion regulators on a cell membrane (blue region). The arrows indicate the movement of ions when the regulators are open. (B). Comparison of neuromuscular excitable and non-excitable somatic cells. Excitable cells usually exhibit action potentials, while the non-excitable somatic cells have membrane potential fluctuations, which vary in their amplitudes and frequencies.
Neuronal and muscular systems have been well investigated for their bioelectric activities. The field of neuromuscular bioelectricity has a relatively long and diverse history [27]. Luigi Galvani first demonstrated the relationship between electricity and animals in 1780 by electrically stimulating frog limbs to cause movement. However, it was almost another hundred years before the first measurements of action potentials, in 1865 by Julius Bernstein, using a differential rheotome [28]. The first intracellular electrical measurements of the resting membrane in the protozoon Paramecium were performed in 1934 [29]. Afterward, ion discoveries on neuronal bioelectricity were made by Hodgkin and Katz, using the giant squid axon as an experimental model [30]. Their intracellular recording studies paved the way for neurology and the fundamental understanding of action potentials [31]. One example is the combinational uses of neuronal axons’ action potential, voltage-gated Ca2+ ion channels, and synaptic neurotransmitters for neural signals [32]. However, the function of bioelectricity remains largely unknown outside of a neuromuscular context. Expanding on these concepts of neuronal bioelectricity and neurotransmitters, it is not inconceivable that other electrical signals could travel across the membranes of non-nerve cells and trigger various responses: to cause other ions to enter the cell (or be released from internal stores); to change transcriptional regulation of the machinery; to cause protein modifications, such as conformation or phosphorylation, to affect function; as well as to modify plasma membrane molecules such as receptors, kinases, and lipids [33,34,35,36][33][34][35][36].
2. Cellular Contributors to Membrane Potential and Bioelectricity
2.1. Cell Membrane Potential and Concentration Gradients
Bioelectricity can be exhibited in several different forms in multicellular organisms: on cellular, tissue, and organ levels. For example, cell membrane potential or membrane voltage (Vm) is one of the integral cellular bioelectric properties (Figure 1A). Many essential cellular physiological processes rely on Vm. These include cross-membrane transport (e.g., nutrients, salts, water), cell volume control, secretion, the cell cycle, and migration [13,25][13][25]. Additionally, Vm allows for cognitive and motor function through neuronal signaling, resulting in organismal, tissue, or cellular sensory detection, and locomotive movement [25]. In typical neuronal signaling, the steady-state baseline voltage is called resting Vm, whereas the excited “signaling” state is called an action potential (AP). Although this generally results in a range between −30 and −80 mV, Vm can even exceed a range of −5 mV to −150 mV, depending on cell type [9]. These resting Vm values can fluctuate in a small or large deviation. Large and rapid depolarization changes from negative to more positive membrane potential are referred to as APs, which are barely reported outside of neuronal and muscular tissues. These APs are triggered by ion channels that respond to changes in voltage that reach a certain threshold. More specifically, depolarizations may merge along a neuron axon or dendrite, eventually pass the Vm threshold for voltage-gated ion channels, and form an AP [39][37]. These AP waves can propagate from multiple locations, and if two meet from opposing directions, they will annihilate each other [40][38]. This quick (millisecond) and extreme (≥100 mV difference) swing in voltage, caused by altering intracellular ion concentrations, is unique to excitatory cells. However, increasing evidence shows smaller and longer-duration types of electrical signaling events in other, non-excitable cell types, such as melanocytes, can have significant effects [41][39]. Changes in Vm of non-excitable somatic cells could come from a variety of factors and would not be classified as traditional AP signals (Figure 1B). Smaller and less extreme increases or decreases in Vm can occur within embryonic neural and non-neural tissues over various periods, such as milliseconds, seconds, minutes, hours, or even days. Such subtle bioelectric signals may be essential in cell differentiation and embryonic patterning during development [7,8,9,37][7][8][9][40]. The electromagnetic force of the differential distribution of ions across the cell membrane generates the electric potential. Thus, the concentration gradient of each ion molecule jointly contributes to Vm value [63][41]. For example, there is a high level of potassium (K+) and low levels of sodium (Na+) within cells at resting Vm. High levels of intracellular K+ and extracellular Na+ ions are mainly established by the sodium/potassium ATPase pump (Figure 1A). One ATPase pump binds three intracellular Na+ ions, utilizes ATP to change conformation via phosphorylation, and releases the three Na+ ions into the extracellular space. Next, two extracellular K+ ions will bind to this outward-facing conformation, causing dephosphorylation and reversal of conformation that allows potassium ions into the cell against its concentration gradient [7,64][7][42]. This form of active transportation and the resulting electrochemical gradient is responsible for high intracellular potassium. The electrical potential difference that counteracts or balances the concentration gradient for a given ion is called equilibrium potential. If only one permeant ion species exists in a cell, its resting membrane potential will equal the equilibrium potential for that ion. Potassium and sodium ions are the two main contributors to membrane potential, but Cl− and Ca2+ ions can also affect Vm, in addition to other charged molecules, such as protons (H+) and organic anions, depending on cell types. Generally, potassium equilibrium potential is close to resting cell membrane potential in many cell types, including glia and neurons. Thus, maintenance of high intracellular potassium is critical for establishing resting Vm [32,63][32][41]. This difference in concentration is hard to maintain, and potassium ions can exit the cell through various leak channels, such as K2P potassium channels on the plasma membrane [65][43]. Removing positively charged K+ ions from the cell will result in a more negative electrical charge, forcing more positive ions to be pulled back into the cell against the chemical gradient. This constant cycling of potassium being pumped into cells and leaking out helps to establish the electric potential of resting Vm. Eventually, these electric and gradient forces will reach equilibrium. This balance can be mathematically described in the Nernst equation [32,63][32][41].2.2. Membrane Potential Contributors: Ion Channels, Gap Junctions, and Solute Carriers
Ion channels are a group of transmembrane proteins that significantly contribute to overall cellular bioelectricity. Channels are essentially small pores in the cell membrane that alter permeability for specific ions based on selectivity (molecular charge and size) and gating (what is required to open the channel) [66][44]. Channel conductivity is aligned with the ion concentration gradient, so energy is not required for a high rate of ion-selective transport. However, the channels will only allow ions to flow down their concentration gradient (moving from high to low concentration areas). The composition of these channels on the cell membrane has been compared to an electronic component called a field-effect transistor [67][45]. In the human genome, more than 400 family members of ion channels are currently characterized, accounting for around 1.5% of the genome [68][46]. A comprehensive list of human ion channel details can be found on the HUGO Gene Nomenclature Committee website and the IUPHAR/BPS Guide to Pharmacology [66,69][44][47]. Based on ion selectivity, ion channels can be classified as sodium (Na+), calcium (Ca2+), potassium (K+), chloride (Cl−), or proton (H+) channels, as well as non-charged molecules such as aquaporins [66,70][44][48]. The most direct Vm-contributing ion channels are K+ and Na+, while the others play a minor role or secondary messenger role, such as that of Ca2+. Each ion channel type can then be further categorized by gating mechanism. One group, voltage-gated channels, will open or close when their voltage-sensitive domains detect a specific change in membrane potential, usually a significant depolarization from action potentials in neurons. Another type, ligand-gated ion channels, relies on their receptor binding a particular ligand to cause or prevent ionic flow. A third category, leak channels, continually allows a small amount of sodium or potassium to leave the cell, regardless of Vm state [65,71,72][43][49][50]. This type of channel can profoundly impact Vm because it can heavily affect the ion gradient at different stages of excitatory conditions. There are additional mechanisms to regulate or gate leak channels, such as temperature, mechanical force, and light [65,71,72][43][49][50]. Another interesting group of channels is that of inwardly rectifying potassium channels (Kir) [73][51]. These channels allow K+ ions to move more easily into, rather than out of, a cell when the cell membrane is depolarized. This is because the intracellular concentration of potassium is so high at rest, and this type of ion movement occurs against the concentration gradient. Even when these are functioning, it is difficult for K+ ions to enter the cell, and they might leak out. Due to this unique characteristic, the Kir channels will impact concentration gradients, resting membrane potential, and cell excitability [73][51]. Furthermore, different channels can show distinct levels of rectification (e.g., high or low). The lipid species, such as PIP2 (phosphatidylinositol 4,5-bisphosphate), can further regulate Kir channels, as can Mg2+, polyamines, phosphorylation, or protein–protein interactions [73][51]. Gap junctions are membrane proteins that physically connect adjacent cells to allow ions, small molecules, and electrical impulses to pass directly by a regulated gate between cells. Like ion channels, their conductance is passive and down an electrochemical gradient. Thus, they do not rely on ATP-like ion pumps. Gap junctions are formed by connecting proteins called connexins and pannexins in vertebrates and innexins in invertebrates (depending on the number of Cys residues in their extracellular loop and glycosylation) [74][52]. These connexins have unique protein structures, properties for permeability, and gating. Each gap junction comprises six connexin subunits on one cell that oligomerize with another six connexins on an adjacent cell. The connection of the same connexin isoform is called homogenous/homomeric, but these properties can change and become more complex by forming heterogeneous/heteromeric gap junctions [75][53]. When these connexins are not coupled to form a gap junction, they are known as hemichannels [76][54]. These hemichannels may serve as an ionic and molecular interchange routes between the cytoplasm and the extracellular environment [77][55]. Gap junctions and hemichannels play significant roles in cell-to-cell communication by exchanging ions, small molecules, subcellular vesicles, electric impulses, and organelles, due to their relatively larger pores [78][56]. Thus, they are natural modulators of cellular bioelectricity. Electrical synapses between neurons can be considered a specialized gap junction. In addition, gap junctions have also been found to be needed for direct cell communication in tunneling nanotubules (TNTs) [79,80][57][58]. Gap junctions are crucial for many physiological processes, including synchronized depolarization of cardiac muscle and embryonic development [81,82][59][60]. One ubiquitous gap junction, connexin 43 (CX43), has been implicated in multiple organisms and diseases and it contributes to electrical signaling [83][61]. Connexin mutations and misregulations have been shown to cause many diseases, such as neurodegenerative diseases and congenital morphological defects in mice and humans [84,85][62][63]. Another group of Vm ion regulators is solute carrier proteins (SLCs). These proteins utilize secondary active transport, where thermodynamically favorable reactions (i.e., ions moving down their concentration gradient) are paired with one or more other molecules to be transported in an unfavorable reaction [86][64]. The free energy provided by the movement in the favorable direction makes movement in the less favorable direction possible and allows transport without directly consuming cellular energy. These reactions utilizing the electrochemical gradient can occur with both substrates moving in the same direction, known as symporters, or substrates moving in opposite directions, known as antiporters. Thus far, over 450 transporter proteins are found in the plasma membrane of cells and subcellular organelles [86,87,88][64][65][66]. These SLCs have an extensive range of substrate specificity, including ions, organic ions, sugars, vitamins, amino acids, nucleotides, oligopeptides, drugs, and metals. In addition, some SLCs can transport multiple different biomolecules, others can only transport a single biomolecule, and up to 30% are “orphan” proteins, whose substrates remain unknown. Thus, these SLCs have been involved in many physiological regulations, such as selective barriers, neurotransmitters, nutrition, and metabolic regulation [86,88][64][66]. More than 190 diseases have been linked to SLCs, such as thyroid, hearing, neurological, metabolic, and congenital defects [86,88][64][66]. Due to the nature of their substrates, the SLCs could be an essential contributor to cellular bioelectricity.3. Bioelectricity Evidence from Zebrafish Genetics
3.1. Zebrafish as a Superior Model for Bioelectric Research
The zebrafish has become one of the leading model organisms used in research since its debut in the 1970s, due to its unique advantages [89,90,91][67][68][69]. First, zebrafish share vertebrate biology with humans. Zebrafish possess 70% orthologous genes to humans [92][70]. Second, it is a relatively affordable model, compared to murine models. Third, small body size and external development make zebrafish embryos an ideal in vivo system. Fourth, tractable genetics has been developed in zebrafish, including large-scale forward genetic mutagenesis, CRISPR-based reverse genetics, and Tol2 transposon-based transgenesis [93,94,95][71][72][73]. Furthermore, a significant source of mutation lines is available through the repository ZFIN, and the greater zebrafish research community is highly collaborative [96,97][74][75]. All these advantages make zebrafish popular for studying developmental biology, neuroscience, physiology, toxicology, drug screens, and many human diseases such as cancers [89,90,91,98,99][67][68][69][76][77]. Zebrafish are also particularly suited to bioelectric research. The combination of excellent and well-established genetic tools with transparent external embryonic development can allow for manageable mutant generation and cutting-edge microscopy to explore previously unattainable information. These attributes can also be useful in bioelectric research. Below, wresearchers highlight bioelectric-related zebrafish studies that demonstrate the importance of this model as an optimal way to characterize and uncover the as-yet-undetermined bioelectric characteristics and mechanistic properties.3.2. Zebrafish Mutants with Adult Fin-Size Change
Zebrafish adults have two sets of fins: paired fins (pectoral and pelvic) and unpaired median fins (dorsal, anal, and caudal), aligning their anterior to posterior body margins [100,101,102][78][79][80]. Each fin comprises endoskeletons and external dermal bones: the fin rays, or lepidotrichs. The adult fin size and its proportion to the body are generally unvarying. Zebrafish paired fin development was reported to share similar mechanisms with tetrapod limbs, as corresponding signaling centers such as ZPA (zone of polarization) and AER (apical ectodermal ridge) were characterized in zebrafish [100,103,104][78][81][82]. Although direct evidence of bioelectricity in zebrafish fin development is still lacking, indirect evidence came from several zebrafish fin mutants from large-scale forward genetic screenings. The first reported zebrafish mutant with elongated fin size is longfin(loft2), which is a dominant mutant that occurred in nature and is present in the widely used Tüpfel fish line. The causal mutant gene of the lof has remained unknown for decades until recently. Two independent reports pinpointed Kcnh2a, a voltage-gated potassium channel [105,106][83][84]. There is a 0.9 Mb chromosomal reversion upstream of the kcnh2a gene on chromosome 2 [106][84]. This inversion disrupts gene regulation and causes a change of the cis-ectopic expression of kcnh2a in zebrafish fins. Similar to the loft2, another longfin (alfdty86d), an ENU-induced mutant, possesses elongated fins in adults in a dominant way [107][85]. This alfdty86d mutant was reported to be caused by gain-of-function mutations in kcnk5b, a potassium leak channel gene [107][85]. The authors also reported larva fish overgrowth and cellular voltage change, indicating the Kcnk5b-mediated bioelectricity of fin anlagen could be the underlying mechanism through local overgrowth [107][85]. The schleier is another zebrafish mutant with elongated fins. This mutation is caused by the inactivation of a potassium–chloride cotransporter, slc12a7a/kcc4a [108][86]. This mutant is also genetically dominant, and homozygous adults exhibit broken stripes and pigmentation alternations. A CRISPR mutation experiment revealed that the function levels of Kcc4a correspond to the fin and barbel lengths. In addition, kcnk5b knockout in the schleier fish embryos can reduce the adult fin lengths, suggesting that Slc12a7a might function together with Kcnk5, and both might be required for bioelectric regulation in wildtype fish. Interestingly, the same research group also identified slc43a2/lat4a, an L-leucine amino acid transporter that can modify the kcnh2a mutation effect in loft2 mutant fish, resulting in a flying-fish-like phenotype [106][84]. This lat4a mutant, lat4anr21, is also dominant and exhibits a short-finned phenotype in heterozygotes. The interactions between Lat4a and Kcnh2a in the flying-fish-like zebrafish suggest they are also involved in bioelectric regulation. Along with this short-finned phenotype, two additional mutants were reported. They are the shortfin (sof) mutants (4 alleles: sofb123 (spontaneous), sofj7e1, sofj7e2, sofj7e3 (ENU-induced)) caused by a hypomorphic mutation in the gap junction, Cx43 [109][87], as well as a fish mutant, mau, caused by a dominant missense mutation in aqp3a (aquaporin 3a) [110][88]. Like the lat4anr21 mutation, the cx43 mutation in sof also reverted the loft2 long-finned phenotype [111][89], suggesting that Cx43 is another bioelectric regulator for zebrafish fin size. OurThe laboratory recently characterized a dominant long-finned mutant, Dhi2059, which was generated via a large-scale insertional mutagenesis [112][90]. The kcnj13 gene’s exon 5 was disrupted by a retroviral insertion. Although this exon encodes 5′ UTR (untranslated region), not protein, viral DNA insertion leads to a transient and ectopic expression of kcnj13 in the somites between 15S (15-somite stage) and 48 dpf (days post fertilization) in Dhi2059 fish embryos. Transgenic fish Tg (−5.4k pax3a: kcnj13-IRES-EGFP), in which the kcnj13 gene is under the control of the −5.4k pax3a promoter, can phenocopy the long-fin phenotype. Thus, kcnj13 misregulation resulted in elongated fins in the adult zebrafish, mainly by increasing the length of fin rays [112][90]. Different from the previously mentioned long-finned mutants (lof, alf, schleier), ourthe results suggest that the adult fin size can be determined at the somite stage in early fish embryos. This indicates that bioelectricity is set up early and could serve as a memory for patterning and size regulation in later ontology (see detailed discussion in the prospective section). In addition, weresearchers showed that transient expression of multiple potassium channels (kcnj1b, kcnj10a, kcnk9, human KCNJ13) in zebrafish early embryos (by microinjection) could also cause chimeric long fins in injected adult fish. This result suggests it is not a specific potassium channel, but that bioelectricity is the key to the elongated fin phenotype. Multiple key points can be obtained by comparing these zebrafish mutants. First, all the mutant genes are involved in ion regulation, which is intrinsically linked to bioelectricity. These ion regulators have their own ion type selecting properties and conductance. It becomes challenging to explain the fin phenotype with a specific channel or ion. Instead, it is more reasonable that electric signaling is the underlying mechanism. Different ion regulators with other properties can be used to construct and modify the bioelectric state of cell groups and tissues. Second, all of these mutations are genetically dominant; most are gain-of-function, ectopically expressed, or neomorphic. Lastly, the specificity of the zebrafish fin-size phenotype may be caused by the spatiotemporal distribution of these ion regulators during embryonic development, as exampled by the Dhi2059 mutant. Taken together, the zebrafish’s adult fin size could be regulated at multiple stages. Although most studies reported altered gene expression in fin anlagen or local fins, ourthe experimental data suggested that somites, the embryonic origin of fin ray progenitor cells, can play a critical patterning role. Consistent with zebrafish mutants, it is also worth noting that different potassium channels were recently identified in other teleosts through genome association studies. The inwardly rectifying channel gene kcnj15 was mapped to long-finned betta fish [113][91]. Additionally, the ether-à-go-go (EAG) potassium channel gene, kcnh8, was found to be highly expressed in the male caudal fins in Xiphophorus [114][92]. Like zebrafish, kcnk5bS was identified as a candidate for long-tailed goldfish [115][93]. Together with zebrafish mutants, these data suggest that ion-channel-mediated bioelectricity plays an essential role in fin patterning.3.3. Zebrafish Mutants with Adult Pigmentation Pattern Alterations
Zebrafish adults exhibit distinct stereotypical stripe patterns along their bodies, with alternating rows of melanophores (dark pigments) and xanthophores (red-orange pigments) mixed with iridophores (iridescent pigments) [116,117,118][94][95][96]. Local and long-range interactions and communication among these different pigment cells during embryonic and larval stages are essential to forming the stripe patterns [79,115,116][57][93][94]. Among many mutant zebrafish lines with altered pigmentation patterns, several are mutations of ion regulators, suggesting that ion-channel-mediated bioelectric signals play important roles in pigmentation patterning. Two of the fish mutants, albino and golden, resulted from the loss of function of solute carrier genes, slc45a2 and slc24a5, respectively [119,120,121][97][98][99]. The phenotypic results of the two mutants are a complete loss of melanophores and light stripes (melanophores with small and fewer melanin granules), respectively. The two genes are expressed in zebrafish melanophores, and light pigmentation was thought to be mainly caused by reduced melanogenesis due to ion and proton alteration in the melanophores [120,121][98][99]. The transparent (tra) fish possess fewer iridophores, melanophores, and dark spots, instead of stripes, in adults. This tra is a loss-of-function mutation of the mpv17 gene, which encodes a non-selective channel that modulates mitochondria membrane potential [122,123][100][101]. Although the loss of Mpv17 was found to cause a reduction in the number of mitochondria and reduced pyrimidine synthesis [123][101], the bioelectricity of iridophores might also contribute to patterning defects. In addition to chromophore defects, zebrafish stripe patterns were found to be altered in additional mutants. The leopard (leot1) mutation, also known as tup, is a spontaneous recessive mutation causing spots in the adult Tüpfel fish line. This mutation is caused by the cx41.8 (connexin 41.8) gene, which encodes Gja5b in zebrafish [124][102]. Similarly, luchs (luctXA9) is a mutation of the cx39.4 (connexin 39.4) gene, which encodes Gja4 [124][102]. Both cx41.8 and cx39.4 are required for melanophore and xanthophore development. Both mutants show aggregated dark spot patterns instead of stripes. Interestingly, it was shown that these two connexins could form heteromeric, in addition to homomeric, gap junctions, which are essential for melanophore and xanthophore cellular communication [124,125][102][103]. Recently, another mutant zebrafish, schleier, was reported to be caused by hypomorphic function of another solute carrier, slc12a7a/kcc4a [108][86]. The homozygous mutant fish show broken stripes in the ventral body flank and anal and caudal fins. Gap junctions usually conduct small molecules and ions between neighboring cells. Thus, they can modulate molecular and electrical coupling among the adjacent cells [81[59][60],82], and over longer distances [79,80][57][58]. Additionally, the obelix(obe)/jaguar(jag) mutants, which are caused by a kcnj13 loss of function, have fewer stripes compared to wildtype fish [126][104]. Kcnj13 is an inwardly rectifying potassium channel that regulates cell excitability and membrane potential. Based on the less severe pigmentation phenotype of the kcnj13 null mutants (kcnj13pu107, kcnj13pu109) ourthe lab generated, the original alleles (jagb230, obetc271d, and obetd15) are most likely dominant negative [112,126][90][104]. More recently, kcnj13 expression was found to underlie the pattern diversification among Danio species via the kcnj13 regulatory changes [127][105]. This potassium channel gene is expressed in melanophores during development, suggesting that it may regulate melanophore bioelectric properties. Indeed, cellular electrical communication was partially disrupted in this mutant. The dissociated melanophores of jag are more depolarized when measured with a voltage-sensitive dye, DiBAC4(3), than the melanophores from wildtype fish. Wildtype melanophores are transiently depolarized when contacted by the dendrites of a xanthophore, and then moved away from the xanthophore. In contrast, jagb230 melanophores lost contact-dependent depolarizations and repulsive migration behavior [128][106]. Three additional zebrafish mutants could also be related to bioelectric regulation, though the related genes are not direct ion regulators. Spermidine is an endogenous polyamine that can regulate ion channels and connexins [129,130][107][108]. The idefix (idet26743) fish is a loss-of-function mutant of the srm (spermidine synthase) gene [131][109]. Homozygous idet26743 mutants have fewer narrowed and often interrupted dark stripes in the trunk and fewer strips in the fins. This ide mutation can further reduce melanophores when crossed with leot1, luctXA9, and obe271d mutants, suggesting that spermidine may modulate connexin and potassium channel functions. Moreover, ectopic expression of spermidine/spermine N1-acetyltransferase (Ssat), a polyamine metabolic enzyme in melanophore, caused broken stripes and a loss of melanophores in the leot1/t1 background, also supporting this idea [132][110]. Another zebrafish mutant, schachbrett (sbrtnh009b), is caused by a loss of function mutation of tight junction protein 1a (Tjp1a), which is expressed in iridophore [133][111]. Like idet1, the sbrtnh009b mutant exhibits more substantial pigment patterning defects in luct32241 and leot1 background, indicating Tjp1a may interact with connexins. Thus, Tjp1a may indirectly affect the bioelectricity of chromatophores. The third zebrafish mutant, mau, also possesses spotted pigments. The underlying gene of the mau mutation is aqp3a, which is mainly expressed in skin and muscle, but not in chromatophores [110][88]. Transplantation of aqp3atVE1/+ blastomere cells into wildtype and Aqp3aR220Q in a transgenic experiment revealed that Aqp3a might indirectly influence chromatophores for pigment patterning. Aqp3a is a transporter of non-polar solutes such as glycerol, peroxide, and urea, excluding ions [134][112]. Thus, Aqp3a can modulate the ion concentrations related to cellular bioelectricity.References
- Lander, A.D. Pattern, growth, and control. Cell 2011, 144, 955–969.
- Salazar-Ciudad, I.; Jernvall, J.; Newman, S.A. Mechanisms of pattern formation in development and evolution. Development 2003, 130, 2027–2037.
- Takahashi, Y.; Osumi, N.; Patel, N.H. Body patterning. Proc. Natl. Acad. Sci. USA 2001, 98, 12338–12339.
- Wolpert, L. Positional information and the spatial pattern of cellular differentiation. J. Theor. Biol. 1969, 25, 1–47.
- Peter, I.S.; Davidson, E.H. Assessing regulatory information in developmental gene regulatory networks. Proc. Natl. Acad. Sci. USA 2017, 114, 5862–5869.
- Negrete, J., Jr.; Oates, A.C. Towards a physical understanding of developmental patterning. Nat. Rev. Genet. 2021, 22, 518–531.
- Levin, M. Molecular bioelectricity: How endogenous voltage potentials control cell behavior and instruct pattern regulation in vivo. Mol. Biol. Cell 2014, 25, 3835–3850.
- Levin, M. Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell 2021, 184, 1971–1989.
- Chang, F.; Minc, N. Electrochemical control of cell and tissue polarity. Annu. Rev. Cell Dev. Biol. 2014, 30, 317–336.
- Levin, M.; Pezzulo, G.; Finkelstein, J.M. Endogenous Bioelectric Signaling Networks: Exploiting Voltage Gradients for Control of Growth and Form. Annu. Rev. Biomed. Eng. 2017, 19, 353–387.
- Mathews, J.; Levin, M. The body electric 2.0: Recent advances in developmental bioelectricity for regenerative and synthetic bioengineering. Curr. Opin. Biotechnol. 2018, 52, 134–144.
- Levin, M. Molecular bioelectricity in developmental biology: New tools and recent discoveries: Control of cell behavior and pattern formation by transmembrane potential gradients. Bioessays 2012, 34, 205–217.
- Kulbacka, J.; Choromanska, A.; Rossowska, J.; Wezgowiec, J.; Saczko, J.; Rols, M.P. Cell Membrane Transport Mechanisms: Ion Channels and Electrical Properties of Cell Membranes. Adv. Anat. Embryol. Cell Biol. 2017, 227, 39–58.
- Catterall, W.A.; Wisedchaisri, G.; Zheng, N. The chemical basis for electrical signaling. Nat. Chem. Biol. 2017, 13, 455–463.
- Stratford, J.P.; Edwards, C.L.A.; Ghanshyam, M.J.; Malyshev, D.; Delise, M.A.; Hayashi, Y.; Asally, M. Electrically induced bacterial membrane-potential dynamics correspond to cellular proliferation capacity. Proc. Natl. Acad. Sci. USA 2019, 116, 9552–9557.
- Chimerel, C.; Field, C.M.; Pinero-Fernandez, S.; Keyser, U.F.; Summers, D.K. Indole prevents Escherichia coli cell division by modulating membrane potential. Biochim. Biophys. Acta 2012, 1818, 1590–1594.
- Wayne, R. The excitability of plant cells: With a special emphasis on characean internodal cells. Bot. Rev. 1994, 60, 265–367.
- Martinac, B.; Saimi, Y.; Kung, C. Ion channels in microbes. Physiol. Rev. 2008, 88, 1449–1490.
- Adamatzky, A. Language of fungi derived from their electrical spiking activity. R. Soc. Open Sci. 2022, 9, 211926.
- Dehshibi, M.M.; Adamatzky, A. Electrical activity of fungi: Spikes detection and complexity analysis. Biosystems 2021, 203, 104373.
- Babikova, Z.; Gilbert, L.; Bruce, T.J.A.; Birkett, M.; Caulfield, J.C.; Woodcock, C.; Pickett, J.A.; Johnson, D. Underground signals carried through common mycelial networks warn neighbouring plants of aphid attack. Ecol. Lett. 2013, 16, 835–843.
- Volkov, A.G.; Shtessel, Y.B. Underground electrotonic signal transmission between plants. Commun. Integr. Biol. 2020, 13, 54–58.
- Strahl, H.; Hamoen, L.W. Membrane potential is important for bacterial cell division. Proc. Natl. Acad. Sci. USA 2010, 107, 12281–12286.
- Eckert, D.; Schulze, T.; Stahl, J.; Rauh, O.; Van Etten, J.L.; Hertel, B.; Schroeder, I.; Moroni, A.; Thiel, G. A small viral potassium ion channel with an inherent inward rectification. Channels 2019, 13, 124–135.
- Abdul Kadir, L.; Stacey, M.; Barrett-Jolley, R. Emerging Roles of the Membrane Potential: Action beyond the Action Potential. Front. Physiol. 2018, 9, 1661.
- Benarroch, J.M.; Asally, M. The Microbiologist’s Guide to Membrane Potential Dynamics. Trends Microbiol. 2020, 28, 304–314.
- Schuetze, S.M. The Discovery of the Action-Potential. Trends Neurosci. 1983, 6, 164–168.
- Carmeliet, E. From Bernstein’s rheotome to Neher-Sakmann’s patch electrode. The action potential. Physiol. Rep. 2019, 7, e13861.
- Kamada, T. Some Observations on Potential Differences across the Ectoplasm Membrane of Paramecium. J. Exp. Biol. 1934, 11, 94–102.
- Hodgkin, A.L.; Katz, B. The effect of sodium ions on the electrical activity of giant axon of the squid. J. Physiol. 1949, 108, 37–77.
- Schwiening, C.J. A brief historical perspective: Hodgkin and Huxley. J. Physiol. 2012, 590, 2571–2575.
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544.
- Maqoud, F.; Curci, A.; Scala, R.; Pannunzio, A.; Campanella, F.; Coluccia, M.; Passantino, G.; Zizzo, N.; Tricarico, D. Cell Cycle Regulation by Ca2+-Activated K+ (BK) Channels Modulators in SH-SY5Y Neuroblastoma Cells. Int. J. Mol. Sci. 2018, 19, 2442.
- Tolstykh, G.P.; Cantu, J.C.; Tarango, M.; Ibey, B.L. Receptor- and store-operated mechanisms of calcium entry during the nanosecond electric pulse-induced cellular response. Biochim. Biophys. Acta BBA-Biomembr. 2019, 1861, 685–696.
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214.
- Zhou, Y.; Wong, C.O.; Cho, K.J.; van der Hoeven, D.; Liang, H.; Thakur, D.P.; Luo, J.; Babic, M.; Zinsmaier, K.E.; Zhu, M.X.; et al. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 2015, 349, 873–876.
- Jan, L.Y.; Jan, Y.N. Voltage-gated potassium channels and the diversity of electrical signalling. J. Physiol. 2012, 590, 2591–2599.
- Fillafer, C.; Paeger, A.; Schneider, M.F. Collision of two action potentials in a single excitable cell. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3282–3286.
- Cervera, J.; Meseguer, S.; Mafe, S. Intercellular Connectivity and Multicellular Bioelectric Oscillations in Nonexcitable Cells: A Biophysical Model. ACS Omega 2018, 3, 13567–13575.
- Harris, M.P. Bioelectric signaling as a unique regulator of development and regeneration. Development 2021, 148, dev180794.
- Wright, S.H. Generation of resting membrane potential. Adv. Physiol. Educ. 2004, 28, 139–142.
- Stone, M.S.; Martyn, L.; Weaver, C.M. Potassium Intake, Bioavailability, Hypertension, and Glucose Control. Nutrients 2016, 8, 444.
- Renigunta, V.; Schlichthörl, G.; Daut, J. Much more than a leak: Structure and function of K2P-channels. Pflüg. Arch. Eur. J. Physiol. 2015, 467, 867–894.
- Alexander, S.P.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Ion channels. Br. J. Pharmacol. 2021, 178 (Suppl. S1), S157–S245.
- Janata, J. Historical review. Twenty years of ion-selective field-effect transistors. Analyst 1994, 119, 2275–2278.
- Hutchings, C.J.; Colussi, P.; Clark, T.G. Ion channels as therapeutic antibody targets. mAbs 2019, 11, 265–296.
- Seal, R.L.; Braschi, B.; Gray, K.; Jones, T.E.M.; Tweedie, S.; Haim-Vilmovsky, L.; Bruford, E.A. Genenames.org: The HGNC resources in 2023. Nucleic Acids Res. 2023, 51, D1003–D1009.
- Finn, R.N.; Cerda, J. Evolution and functional diversity of aquaporins. Biol. Bull. 2015, 229, 6–23.
- Cochet-Bissuel, M.; Lory, P.; Monteil, A. The sodium leak channel, NALCN, in health and disease. Front. Cell. Neurosci. 2014, 8, 132.
- Enyedi, P.; Czirjak, G. Molecular background of leak K+ currents: Two-pore domain potassium channels. Physiol. Rev. 2010, 90, 559–605.
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 2010, 90, 291–366.
- Scemes, E.; Spray, D.C.; Meda, P. Connexins, pannexins, innexins: Novel roles of “hemi-channels”. Pflug. Arch. Eur. J. Physiol. 2009, 457, 1207–1226.
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204.
- Cottrell, G.T.; Burt, J.M. Functional consequences of heterogeneous gap junction channel formation and its influence in health and disease. Biochim. Biophys. Acta BBA-Biomembr. 2005, 1711, 126–141.
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400.
- Adams, D.S.; Levin, M. Endogenous voltage gradients as mediators of cell-cell communication: Strategies for investigating bioelectrical signals during pattern formation. Cell Tissue Res. 2013, 352, 95–122.
- Eom, D.S.; Bain, E.J.; Patterson, L.B.; Grout, M.E.; Parichy, D.M. Long-distance communication by specialized cellular projections during pigment pattern development and evolution. eLife 2015, 4, e12401.
- Sherer, N.M.; Mothes, W. Cytonemes and tunneling nanotubules in cell–cell communication and viral pathogenesis. Trends Cell Biol. 2008, 18, 414–420.
- Mese, G.; Richard, G.; White, T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2516–2524.
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388.
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girão, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630.
- Lecanda, F.; Warlow, P.M.; Sheikh, S.; Furlan, F.; Steinberg, T.H.; Civitelli, R. Connexin43 Deficiency Causes Delayed Ossification, Craniofacial Abnormalities, and Osteoblast Dysfunction. J. Cell Sci. 2000, 151, 931–944.
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta BBA-Biomembr. 2018, 1860, 192–201.
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A guide to plasma membrane solute carrier proteins. FEBS J. 2021, 288, 2784–2835.
- Alexander, S.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Southan, C.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Transporters. Br. J. Pharmacol. 2021, 178 (Suppl. S1), S412–S513.
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug. Discov. 2015, 14, 543–560.
- Grunwald, D.J.; Eisen, J.S. Headwaters of the zebrafish—Emergence of a new model vertebrate. Nat. Rev. Genet. 2002, 3, 717–724.
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367.
- Liu, S.; Leach, S.D. Zebrafish models for cancer. Annu. Rev. Pathol. 2011, 6, 71–93.
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503.
- Li, M.; Zhao, L.; Page-McCaw, P.S.; Chen, W. Zebrafish Genome Engineering Using the CRISPR-Cas9 System. Trends Genet. 2016, 32, 815–827.
- Lawson, N.D.; Wolfe, S.A. Forward and reverse genetic approaches for the analysis of vertebrate development in the zebrafish. Dev. Cell 2011, 21, 48–64.
- Kawakami, K. Tol2: A versatile gene transfer vector in vertebrates. Genome Biol. 2007, 8 (Suppl. S1), S7.
- Sprague, J.; Clements, D.; Conlin, T.; Edwards, P.; Frazer, K.; Schaper, K.; Segerdell, E.; Song, P.; Sprunger, B.; Westerfield, M. The Zebrafish Information Network (ZFIN): The zebrafish model organism database. Nucleic Acids Res. 2003, 31, 241–243.
- Sprague, J.; Doerry, E.; Douglas, S.; Westerfield, M. The Zebrafish Information Network (ZFIN): A resource for genetic, genomic and developmental research. Nucleic Acids Res. 2001, 29, 87–90.
- Santoriello, C.; Zon, L.I. Hooked! Modeling human disease in zebrafish. J. Clin. Investig. 2012, 122, 2337–2343.
- Veldman, M.B.; Lin, S. Zebrafish as a Developmental Model Organism for Pediatric Research. Pediatr. Res. 2008, 64, 470–476.
- Grandel, H.; Schulte-Merker, S. The development of the paired fins in the zebrafish (Danio rerio). Mech. Dev. 1998, 79, 99–120.
- Pfefferli, C.; Jazwinska, A. The art of fin regeneration in zebrafish. Regeneration 2015, 2, 72–83.
- Mabee, P.M.; Crotwell, P.L.; Bird, N.C.; Burke, A.C. Evolution of median fin modules in the axial skeleton of fishes. J. Exp. Zool. 2002, 294, 77–90.
- Mari-Beffa, M.; Murciano, C. Dermoskeleton morphogenesis in zebrafish fins. Dev. Dyn. 2010, 239, 2779–2794.
- Yano, T.; Abe, G.; Yokoyama, H.; Kawakami, K.; Tamura, K. Mechanism of pectoral fin outgrowth in zebrafish development. Development 2012, 139, 2916–2925.
- Stewart, S.; Le Bleu, H.K.; Yette, G.A.; Henner, A.L.; Robbins, A.E.; Braunstein, J.A.; Stankunas, K. longfin causes cis-ectopic expression of the kcnh2a ether-a-go-go K+ channel to autonomously prolong fin outgrowth. Development 2021, 148, dev199384.
- Daane, J.M.; Blum, N.; Lanni, J.; Boldt, H.; Iovine, M.K.; Higdon, C.W.; Johnson, S.L.; Lovejoy, N.R.; Harris, M.P. Modulation of bioelectric cues in the evolution of flying fishes. Curr. Biol. 2021, 31, 5052–5061.e8.
- Perathoner, S.; Daane, J.M.; Henrion, U.; Seebohm, G.; Higdon, C.W.; Johnson, S.L.; Nüsslein-Volhard, C.; Harris, M.P. Bioelectric Signaling Regulates Size in Zebrafish Fins. PLoS Genet. 2014, 10, e1004080.
- Lanni, J.S.; Peal, D.; Ekstrom, L.; Chen, H.; Stanclift, C.; Bowen, M.E.; Mercado, A.; Gamba, G.; Kahle, K.T.; Harris, M.P. Integrated K+ channel and K+Cl− cotransporter functions are required for the coordination of size and proportion during development. Dev. Biol. 2019, 456, 164–178.
- Iovine, M.K.; Higgins, E.P.; Hindes, A.; Coblitz, B.; Johnson, S.L. Mutations in connexin43 (GJA1) perturb bone growth in zebrafish fins. Dev. Biol. 2005, 278, 208–219.
- Eskova, A.; Chauvigné, F.; Maischein, H.-M.; Ammelburg, M.; Cerdà, J.; Nüsslein-Volhard, C.; Irion, U. Gain-of-function mutations in Aqp3a influence zebrafish pigment pattern formation through the tissue environment. Development 2017, 144, 2059–2069.
- Iovine, M.K.; Johnson, S.L. Genetic analysis of isometric growth control mechanisms in the zebrafish caudal Fin. Genetics 2000, 155, 1321–1329.
- Silic, M.R.; Wu, Q.; Kim, B.H.; Golling, G.; Chen, K.H.; Freitas, R.; Chubykin, A.A.; Mittal, S.K.; Zhang, G. Potassium Channel-Associated Bioelectricity of the Dermomyotome Determines Fin Patterning in Zebrafish. Genetics 2020, 215, 1067–1084.
- Zhang, W.; Wang, H.; Brandt, D.Y.C.; Hu, B.; Sheng, J.; Wang, M.; Luo, H.; Li, Y.; Guo, S.; Sheng, B.; et al. The genetic architecture of phenotypic diversity in the Betta fish (Betta splendens). Sci. Adv. 2022, 8, eabm4955.
- Schartl, M.; Kneitz, S.; Ormanns, J.; Schmidt, C.; Anderson, J.L.; Amores, A.; Catchen, J.; Wilson, C.; Geiger, D.; Du, K.; et al. The Developmental and Genetic Architecture of the Sexually Selected Male Ornament of Swordtails. Curr. Biol. 2021, 31, 911–922.e4.
- Kon, T.; Omori, Y.; Fukuta, K.; Wada, H.; Watanabe, M.; Chen, Z.; Iwasaki, M.; Mishina, T.; Matsuzaki, S.S.; Yoshihara, D.; et al. The Genetic Basis of Morphological Diversity in Domesticated Goldfish. Curr. Biol. 2020, 30, 2260–2274.e6.
- Singh, A.P.; Nüsslein-Volhard, C. Zebrafish Stripes as a Model for Vertebrate Colour Pattern Formation. Curr. Biol. 2015, 25, R81–R92.
- Patterson, L.B.; Parichy, D.M. Zebrafish Pigment Pattern Formation: Insights into the Development and Evolution of Adult Form. Annu. Rev. Genet. 2019, 53, 505–530.
- McGowan, K.A.; Barsh, G.S. How the zebrafish got its stripes. eLife 2016, 5, e14239.
- Dooley, C.M.; Schwarz, H.; Mueller, K.P.; Mongera, A.; Konantz, M.; Neuhauss, S.C.; Nusslein-Volhard, C.; Geisler, R. Slc45a2 and V-ATPase are regulators of melanosomal pH homeostasis in zebrafish, providing a mechanism for human pigment evolution and disease. Pigment. Cell Melanoma Res. 2013, 26, 205–217.
- Lamason, R.L.; Mohideen, M.A.; Mest, J.R.; Wong, A.C.; Norton, H.L.; Aros, M.C.; Jurynec, M.J.; Mao, X.; Humphreville, V.R.; Humbert, J.E.; et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 2005, 310, 1782–1786.
- Le, L.; Escobar, I.E.; Ho, T.; Lefkovith, A.J.; Latteri, E.; Haltaufderhyde, K.D.; Dennis, M.K.; Plowright, L.; Sviderskaya, E.V.; Bennett, D.C.; et al. SLC45A2 protein stability and regulation of melanosome pH determine melanocyte pigmentation. Mol. Biol. Cell 2020, 31, 2687–2702.
- D’Agati, G.; Beltre, R.; Sessa, A.; Burger, A.; Zhou, Y.; Mosimann, C.; White, R.M. A defect in the mitochondrial protein Mpv17 underlies the transparent casper zebrafish. Dev. Biol. 2017, 430, 11–17.
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The Human Mitochondrial DNA Depletion Syndrome Gene MPV17 Encodes a Non-selective Channel That Modulates Membrane Potential. J. Biol. Chem. 2015, 290, 13840–13861.
- Irion, U.; Frohnhöfer, H.G.; Krauss, J.; Çolak Champollion, T.; Maischein, H.-M.; Geiger-Rudolph, S.; Weiler, C.; Nüsslein-Volhard, C. Gap junctions composed of connexins 41.8 and 39.4 are essential for colour pattern formation in zebrafish. eLife 2014, 3, e05125.
- Watanabe, M.; Sawada, R.; Aramaki, T.; Skerrett, I.M.; Kondo, S. The Physiological Characterization of Connexin41.8 and Connexin39.4, Which Are Involved in the Striped Pattern Formation of Zebrafish. J. Biol. Chem. 2016, 291, 1053–1063.
- Iwashita, M.; Watanabe, M.; Ishii, M.; Chen, T.; Johnson, S.L.; Kurachi, Y.; Okada, N.; Kondo, S. Pigment Pattern in jaguar/obelix Zebrafish Is Caused by a Kir7.1 Mutation: Implications for the Regulation of Melanosome Movement. PLoS Genet. 2006, 2, e197.
- Podobnik, M.; Frohnhofer, H.G.; Dooley, C.M.; Eskova, A.; Nusslein-Volhard, C.; Irion, U. Evolution of the potassium channel gene Kcnj13 underlies colour pattern diversification in Danio fish. Nat. Commun. 2020, 11, 6230.
- Inaba, M.; Yamanaka, H.; Kondo, S. Pigment Pattern Formation by Contact-Dependent Depolarization. Science 2012, 335, 677.
- Williams, K. Interactions of polyamines with ion channels. Biochem. J. 1997, 325 Pt 2, 289–297.
- Nichols, C.G.; Lee, S.J. Polyamines and potassium channels: A 25-year romance. J. Biol. Chem. 2018, 293, 18779–18788.
- Frohnhofer, H.G.; Geiger-Rudolph, S.; Pattky, M.; Meixner, M.; Huhn, C.; Maischein, H.M.; Geisler, R.; Gehring, I.; Maderspacher, F.; Nusslein-Volhard, C.; et al. Spermidine, but not spermine, is essential for pigment pattern formation in zebrafish. Biol. Open 2016, 5, 736–744.
- Watanabe, M.; Watanabe, D.; Kondo, S. Polyamine sensitivity of gap junctions is required for skin pattern formation in zebrafish. Sci. Rep. 2012, 2, 473.
- Fadeev, A.; Krauss, J.; Frohnhofer, H.G.; Irion, U.; Nusslein-Volhard, C. Tight Junction Protein 1a regulates pigment cell organisation during zebrafish colour patterning. eLife 2015, 4, e06545.
- Verkman, A.S. Aquaporins at a glance. J. Cell Sci. 2011, 124, 2107–2112.
More