1. Introduction
Lipoprotein(a) (Lp(a)) was first described by K. Berg in 1963
[1]. For several years, interest in this lipoprotein was modest, until the sequencing of the
LPA gene encoding apolipoprotein(a) (apo(a)), a key constituent of Lp(a)
[2], and the subsequent recognition of Lp(a) as an independent risk factor for cardiovascular disease by epidemiological and Mendelian randomization studies
[3][4][3,4]. Later, high Lp(a) values were also associated with calcific aortic valve stenosis
[5] and increased risk of ischemic stroke
[6][7][6,7].
2. Structure and Features of Lp(a)
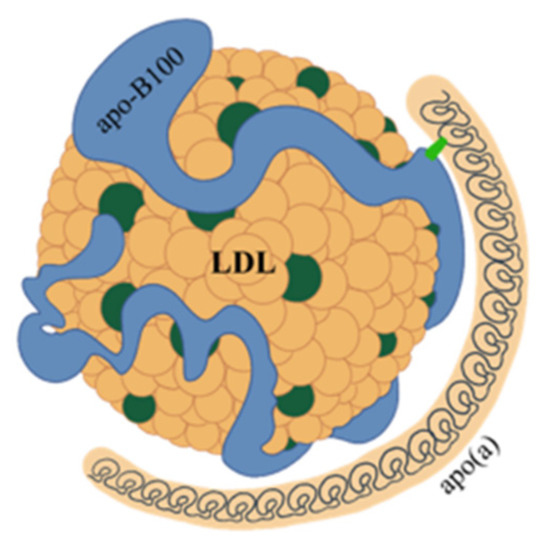
Lp(a) consists of a very low-density lipoprotein (LDL)-like particle with apoprotein B100 being bound via a single disulfide link to a single apoprotein(a) (apo(a)) (Figure 1).
Figure 1. Structure of lipoprotein(a). Lipoprotein(a) consists of a very low-density lipoprotein (LDL)-like particle, with apoprotein B100 (apo-B100) being bound via a single disulfide link to a single apoprotein a (apo(a)).
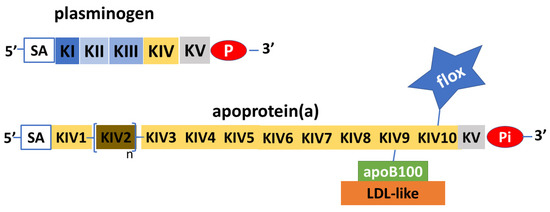
Apo(a) is a glycoprotein that has structural similarities with plasminogen. Plasminogen is made from a protein chain with a terminal part with protease activity joined to five aggregates of about 90 amino acids that have been named kringles because of their shape resembling that of a typical Northern European cake. In plasminogen, the kringles are present in single copy and are named with Roman numerals from KI to KV. Unlike plasminogen, apo(a) has an inactive protease domain and does not contain KI, KII, or KIII kringles, but consists of only one KV subtype and ten KIV subtypes, which are present in single copy except for the KIV
2 subtype that can repeat from one to forty times
[8] (
Figure 2). Polypeptide structures similar to kringle domains are found in several enzymes (thrombin, tissue plasminogen activator, hepatocyte growth factor) as well as in plasminogen and Lp(a). The KVs of plasminogen and apo(a) have strong structural homology, as do the KIVs of plasminogen and Lp(a). However, KV and KIV are quite different from each other, both structurally and as amino acid composition. In contrast, the various subtypes 1 to 10 of KIV have important structural homologies and differ in their composition by only a few amino acids. These small differences may, however, account for different activities such as, for example, the ability to create disulfide bonds between KIV9 and apoB100. The specific function of KIV2 is not known, nor is the physiological function of Lp(a). The characteristic of KIV2 is its ability to be replicated a highly variable number of times in the structure of apo(a)
[9].
Figure 2. Schematic depiction of plasminogen and apoprotein (a). Apoprotein(a) is a glycoprotein with remarkable similarities to plasminogen, in both the protease part (P) and the inactive part (Pi), and in the kringle protein structure (K). The apoprotein(a) does not have KI to KIII, while it has KIV to KV; KV and subtypes 1 and 3 to 10 are expressed in single copy, while subtype 2 is pre-sent in variable copy numbers. Apoprotein(a) can vary greatly in length and molecular weight. Sialic acid (SA) with other N-glucans and O-glucans has structural functions and protects apoprotein(a) from proteases. Apoprotein(a) transports a considerable amount of oxidized phospholipids (flox), which, added to the LDL-like ones connected to apoprotein(a) via apoprotein B100 (ApoB100), make lipoprotein(a) the major carrier of oxidized phospholipids.
Thus, the length of apo(a) is highly variable and its molecular weight has a range from 275 to 800 kDa. Apo(a) contains a fair amount of oligosaccharides, in particular, sialic acid and other N-glycans and O-glycans, which have structural and protective functions against the activity of proteolytic enzymes
[10][11][10,11]. Another relevant aspect in the structure of apo(a) is the presence of oxidized phospholipids, mainly attached to the KIV
10 subtype. Considering also the proportion contained in the LDL-like component, Lp(a) is the major carrier of oxidized phospholipids
[12], so individuals with high Lp(a) values are at greater risk of coronary heart disease and stroke. Apo(a) is produced exclusively by the endoplasmic reticulum of the liver, where it also undergoes post-translational modifications with the creation of a disulfide bond in each kringle that assumes the characteristic shape. The formation process is completed in the Golgi apparatus where the lipoprotein undergoes glycosylation
[13]. The question of whether the assembly of Lp(a) with apo(a) and apoprotein B-100 takes place in the hepatocyte, on the surface or even outside, is not settled yet. Some studies suggest that an initial non-covalent bond between apo(a) and apoprotein B100 could be formed inside the hepatocyte
[10], while the final disulfide bond that completes the formation of Lp(a) would be created outside the hepatocyte, in the Disse spaces of the hepatic sinusoids
[14][15][14,15]. A small proportion of apo(a) remains unaggregated and is eliminated with urine; no particular function has been attributed to it
[16]. The amount of circulating Lp(a) depends on how much apo(a) is produced by the liver, which, in turn, depends on the activity of the
LPA gene, which, being made up of two codominant alleles, encodes two isoforms of apo(a) that may differ in length. In each individual, therefore, there are two different Lp(a)s which may have different molecular weights. The variability in Lp(a) levels in the general population is extremely high, and its plasma concentration can range from 0.1 to 300 mg/dL. Hepatic production of Lp(a) is 80–90% genetically determined; therefore, it is little affected by environmental factors and tends to be stable in adults
[17]. Therefore, in this age group, a single Lp(a) assay may be sufficient to know the related risk profile of the individual. However, this point is a matter of debate, as it is reported that Lp(a) levels would change over time
[18]; moreover, this occurs in the presence of certain diseases. First, it should be noted that Lp(a) behaves as an acute phase protein, so it tends to increase in inflammatory states
[19] and this should be taken into account when scheduling the measurement. In addition, nephropathies are also associated with significant increases in Lp(a) levels, which can be explained by different mechanisms depending on the type of kidney disease. Already in the early stages of renal failure, an increase in Lp(a) can be shown, with increases proportional to the decrease in glomerular filtrate. However, in the nephrotic syndrome, Lp(a) can increase up to fourfold due to an increment of its synthesis by the liver
[20]. Chronic kidney disease patients have a significantly increased risk of cardiovascular disease, related to the level of Lp(a)
[21]. The amount of circulating Lp(a) is inversely proportional to the length of the apo(a), thus to the number of KIV
2 kringles. Therefore, individuals with longer apo(a) isoforms of Lp(a), thus with higher molecular weight, produce less Lp(a) than those with shorter, lower molecular weight isoforms. The lower amount of Lp(a) produced by individuals in which the higher molecular weight isoforms predominate could be due to the longer time required for its production
[22] or to the greater molecular weight isoforms that have not assumed the correct conformation being translocated outside the endoplasmic reticulum, ubiquitinated and degraded
[23]. In addition to length, other genetic factors may influence the increased or decreased production of Lp(a). In particular, several single nucleotide polymorphisms of the
LPA gene are known to be associated with increased or decreased apo(a) and Lp(a) production
[24][25][24,25]. Regarding cardiovascular risk, it is currently believed that it would be related to the amount of circulating Lp(a) and not to its qualitative characteristics. Thus, subjects with higher plasma concentrations of this lipoprotein, who are those with lower molecular weight isoforms of Lp(a), are most at risk
[3]. Lp(a) is metabolized mainly by the liver and to a lesser extent by the kidney. Several receptors are called upon for Lp(a) uptake, such as the BI scavenger receptor, plasminogen receptors, and LDL receptors, but these aspects are not yet fully elucidated
[26]. The variability of Lp(a) gives reason for the difficulties there are in developing precise laboratory methods for its assay. In particular, when polyclonal antibodies, also directed toward the repeated part of apo(a) (KIV
2), are used, larger isoforms tend to be overestimated, while smaller ones may be underestimated
[27]. These difficulties can be overcome by performing analyses with calibrators containing Lp(a) of different lengths or with monoclonal antibodies directed only toward the single-copy portion of apo(a). Lp(a) assays are often expressed in mg/dL, as if directly measuring the entire lipoprotein, and thus, not only its protein part but also its cholesterol content, cholesterol esters, phospholipids, triglycerides, and carbohydrates. This is not quite correct because the assays reflect the quantitative molar ratio between apo(a) and the antibodies that interact with it. Therefore, it would be more correct to express Lp(a) values in nmol/L
[27]. Again because of the variability of Lp(a), there can be no precise conversion factor between mg/dL and nmol/L. However, from a practical point of view, a conversion factor between 2 and 2.5 is still accepted
[28].
3. Lp(a) and Cardiovascular Risk
Lp(a) is associated with an increased risk of cardiovascular events because of some of its structural and functional characteristics that promote atherosclerotic plaque formation and growth. In addition, it has been suggested that Lp(a) promotes coagulation processes following plaque rupture. This, however, is currently only a hypothesis and needs validation. Lp(a) contains a proportion of cholesterol that corresponds to about 30–40% of its weight
[29]. This amount of cholesterol is less than that found in LDL and is negligible when circulating Lp(a) is in low concentration. However, in individuals who have high Lp(a) values, its contribution to the amount of atherogenic cholesterol can be significant, mainly due to the ease with which Lp(a) crosses the endothelium and accumulates in the inner layer of the artery wall, where it is able to activate smooth muscle cell proliferation and migration and foam cell formation
[13]. Interestingly, high levels of Lp(a) promote the earlier stages of atherosclerosis and not just the more advanced phases. Imaging studies have shown that Lp(a) promotes the development of arterial wall inflammation
[30] and that in the presence of coronary artery disease, high Lp(a) levels are associated with an increase in the amount of calcium and necrotic core volume from the atherosclerotic plaque
[31][32][31,32]. The processes promoting inflammation and calcium deposition seem mainly related to the presence of oxidized phospholipids
[33][34][33,34], which would be responsible for the secretion of chemotactic substances and proinflammatory cytokines, upregulation of adhesion molecules, and trans-endothelial migration of monocytes
[30][35][30,35]. Finally, the presence in apo(a) of a protease-like domain similar to that of plasminogen, but inactive, suggests that Lp(a) might also have a role in promoting coagulation processes and reducing fibrinolysis. The latter effect of Lp(a) is still under debate
[36]. However, it seems that Lp(a) has no role in venous thromboembolism events
[37]. In summary, Lp(a) would have pro-atherosclerotic, pro-inflammatory, and possibly pro-coagulant and antifibrinolytic effects. Regarding calcific aortic valve stenosis, numerous studies have shown micro- and macro-calcifications of the aortic valve in adults aged 45 to 55 years with elevated Lp(a) values
[5][38][39][5,38,39]. It is estimated that individuals with higher Lp(a) percentiles are three times more likely to develop calcific aortic valve stenosis than those with lower Lp(a) percentiles
[38]. Furthermore, high plasma Lp(a) levels correlate with more rapid progression of valve stenosis
[12]. The etiopathogenetic mechanisms explaining the association between Lp(a) and calcific aortic valve stenosis have yet to be elucidated; however, oxidized phospholipids would again be called into play, which, in addition to activating inflammatory processes, would stimulate the activation of genes that regulate osteoblastic processes in the cells of the valve interstitium
[12][35][12,35].
Some considerations should be added regarding the relationship between hypolipidemic drug therapy and the levels of Lp(a). Statins have been reported to be associated with a tendency to increase Lp(a) levels, whereas ezetimibe would have no effect
[40][41][40,41].
In contrast, apheresis
[42] and drugs such as niacin
[43] and PCSK9 inhibitors
[44] are able to reduce plasma Lp(a) concentrations. However, while it has been shown that the decrease in Lp(a) levels induced by apheresis is approximately 75% and is associated, in subjects with extremely high Lp(a) levels, with a significant reduction in cardiovascular risk
[42], the same clinical outcome is not achieved with the administration of niacin or PCSK9 inhibitors
[45][46][45,46], which induce reductions in Lp(a) values of 20–30%
[43][44][43,44]. On the basis of the current data, a significant reduction in cardiovascular risk by lowering Lp(a) awaits further studies. Of note, niacin and PCSK9 inhibitors do not currently have a pediatric indication.
4. Lp(a) in Adults
The plasma concentration of Lp(a) is widely variable, and in all populations, there are individuals with very-high Lp(a) values. However, important differences can be found in the distribution of Lp(a) in different ethnic groups. In fact, average Lp(a) values increase from Chinese, to South Asians, to Whites to Blacks, with average concentrations of 16, 19, 31, and 75 nmol/L, respectively
[47]. Hispanics have values intermediate between South Asians and Caucasian Whites
[48]. In general then, Black individuals are those with higher Lp(a) values; in fact, the mean value of the plasma Lp(a) concentration among Blacks is in the fifth quintile of the White population distribution
[49]. These differences among the different ethnic groups would be largely explained by the greater presence of isoforms with lower KIV
2 kringle number in the Black, Caucasian, and Hispanic populations, respectively, although mutations in the
LPA gene and other yet unknown factors would play a non-secondary role
[50][51][50,51]. A recent large study confirms the inverse relationship between the distribution of Lp(a) values and its isoforms and the different distribution among different ethnic groups
[52]. The study shows that Chinese and South Asian populations have the lowest average Lp(a) values and largest isoforms, while Africans and Arabs have the highest concentrations and smallest isoforms. Europeans, Latin Americans, and Southeast Asians rank in between for both parameters. The same study shows a correlation between the presence of Lp(a) values greater than 50 mg/dL with the risk of myocardial infarction with an odds ratio of 1.48 (95% CI: 1.32–1.67) in the entire population. The risk increased from Africans, to Arabs, Chinese, Europeans, Latin Americans to Southeast Asians who had the highest odds ratio
[52]. This study suggests that ethnicity should also be taken into account when assessing the cardiovascular risk associated with Lp(a) levels, as similar Lp(a) values might be associated with different patterns of cardiovascular risk in different ethnicities. Lp(a) values could also differ according to gender. However, the reports on this issue are conflicting: some authors report that males have higher values than females
[53], while for other authors, the opposite would be true
[47]. In males, Lp(a) values would remain consistent throughout adult life
[54], whereas in females, there would be an increase after menopause
[55][56][57][58][55,56,57,58]. In adults, the correlation between Lp(a) levels and cardiovascular risk has a linear and continuous pattern. In general, individuals with Lp(a) values less than 30 mg/dL can be considered at low risk and those with values greater than 50 mg/dL at high risk. Concentrations between 30 and 50 mg/dL are considered borderline
[27]. It should be emphasized, however, that in the assessment of an individual’s cardiovascular risk, Lp(a) values are only one aspect, albeit an important one, that must be considered in the totality of the other known risk factors. The most recent European guidelines suggest that Lp(a) should be measured at least once in a lifetime in adults and that this information should be included in the overall estimate of the risk of developing atherosclerotic-based disease
[27].