Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by José Ignacio Erices.
Glioblastoma (GBM) is the most common and malignant primary brain cancer in adults. Without treatment the mean patient survival is approximately 6 months, which can be extended to 15 months with the use of multimodal therapies. The low effectiveness of GBM therapies is mainly due to the tumor infiltration into the healthy brain tissue, which depends on GBM cells’ interaction with the tumor microenvironment (TME). The interaction of GBM cells with the TME involves cellular components such as stem-like cells, glia, endothelial cells, and non-cellular components such as the extracellular matrix, enhanced hypoxia, and soluble factors such as adenosine, which promote GBM’s invasiveness.
- glioblastoma
- glioblastoma stem-like cells
- tumor microenvironment
1. Introduction
Glioblastoma (GBM) is the most frequent and deadly malignant brain tumor in adults, with an estimated incidence in the United States of 3–5 cases per 100,000 person-years [1]. Currently, GBM treatment consists of multimodal therapy, which includes surgery, radio- and chemotherapy, temozolomide (TMZ) being the gold standard drug [2]. Despite multimodal therapy, the average patient survival does not exceed 15 months and only 26% of patients live up to 2 years post-treatment [2]. The low efficacy of GBM therapies is mainly caused by tumor cell infiltration into the healthy brain tissue, which makes the total surgical tumor resection challenging, impairs the localized effect of radiotherapy and leads inevitably to GBM’s recurrence with fatal outcomes [3]. The GBM infiltration phenotype is considered one of the main characteristics associated with the therapy failure, so understanding its cellular and molecular mechanisms will help to develop new therapeutic strategies to decrease GBM’s aggressiveness. GBM cells are found in particular niches, characterized by extracellular matrix (ECM) interaction, low oxygen pressure, tumor-associated soluble factors and different cell types composing the tumor microenvironment (TME), which is strongly linked to the modulation of the GBM invasive phenotype [4,5][4][5].
2. Cellular Invasion Mechanisms
Despite non-brain tumor cells invading organs through the circulatory system or lymph, GBM cells are considered non-metastatic to extra-cranial organs; however, they perform local invasion into the healthy brain tissues [6]. GBM cells are able to infiltrate through perivascular space around blood vessels or between neurons and glia [7,8][7][8]. The cell infiltration into the brain parenchyma requires the activation of cellular processes such as interaction and degradation of components of the ECM, remodeling of the cytoskeleton, and changes in cell volume [7]. Various tumor cell movement patterns have been described, all of which allow the cells to invade the surrounding tissue in a single-cell movement manner, which comprises mesenchymal or amoeboid movement, and collective movement by the so-called “cluster or strand” [9]. Single-cell movement is the main cellular infiltration mechanism responsible for tumor recurrence and, surprisingly, the same movement pattern is the one used by neural stem cells (NSCs) during the embryonic nervous system development and the response to brain tissue damage [10]. In recent years, emerging evidence has suggested that GBM cell invasion mechanisms are not only dependent on inherent cell characteristics but are also strongly regulated by dynamic communications and interactions between tumor cells and their TME [11].3. Tumor Microenvironment
Most aggressive tumors, such as GBM, can modify their microenvironment, favoring the development of tumorigenic properties, such as chemoresistance, cell proliferation, migration, and invasion [11]. GBM TME components are both cellular (e.g., glioblastoma stem-like cells, endothelial cells, microglia, astrocytes, and neurons) and non-cellular (e.g., ECM, variations in hypoxia levels and soluble molecules) [12].3.1. Cellular Components Involved in GBM Cell Invasion
3.1.1. Glioblastoma Stem-Like Cells
One feature of GBM is its high cellular heterogeneity, which has been used as a prognostic indicator [13]. The presence of cells with self-renewing and multi-lineage differentiation properties called glioblastoma stem-like cells (GSCs), has been proposed as the main cause of tumor initiation, growth, and recurrence during the progression of GBM [14]. Nowadays, GSCs are identified with the use of biomarkers associated with the NSC membrane protein, such as CD133 and CD44 [14]. GBM cell subtypes enriched with CD133 display an increased migratory and invasive capacity, resembling molecular profiles described for angiogenesis and cell invasion [15]. Yu et al. showed that GSCs CD133(+) have a more migratory and invasive phenotype than GSCs CD133(−) both in vitro and in vivo [16]. Organotypic rat brain slices inoculated with GBM CD133(+) cells showed a greater infiltration of healthy brain tissue, mainly through perivascular niches and white matter tracts, than GBM CD133(−) cells [16]. Nishikawa et al. reported CD44 as another GBM biomarker in tumors with exacerbated migration and invasion pathways, preferentially at the tumor periphery [17]. CD44 knockdown inhibited GSCs’ migration and invasion both in vitro and in vivo [17]; mouse brain tumors generated from CD44-knockdown GSCs were less invasive and mice survived significantly longer than control mice [18]. Furthermore, Cheray et al. identified that KLRC3, the gene coding for NK cell group 2 isoform E (NKG2E) protein, is overexpressed in glioblastoma undifferentiated cells compared to the differentiated ones and that its silencing decreases the GSCs’ invasion [19]. Other groups have reported an upregulation of proteins involved in the migration and invasion of GSCs, such as matrix metalloproteinases (MMPs), different members of adamalysins including ADAMs (A disintegrin and metalloproteases) and ADAMTS (A disintegrin-like and metalloprotease domain (reprolysin type) with thrombospondin type 1 motifs), and adhesion receptor proteins (integrins) [20]. Alonso et al. observed that the ectopic expression of the transcription factor SOX2 was essential to induce and maintain GSCs’ migration and invasion [21]. In addition, researchers have measured high expression levels of stem cell markers in the tumor frontal invasion border, supporting the idea of GSCs as being responsible for the highly invasive phenotype of GBM [22]. Several signaling pathways, such as Wnt (wingless-INT), TGF-β (transforming growth factor-beta) and hedgehog (Hh)-GLI1 (glioma-associated oncogene homolog 1) pathways have been reported as determinant modulators of the GBM invasive phenotype [23]. Impaired Wnt signaling is related to GBM’s poor prognosis and its aberrant activation is implicated in the maintenance of a highly invasive phenotype [24]. The intranuclear localization of β-catenin has been reported at the infiltrating edge of tumors while the activation of the canonical Wnt/β-catenin pathway maintains the stemness and enhances GSCs’ mobility through ZEB1 (zinc finger E-box-binding homeobox 1) activation [25]. Likewise, Wnt5a, a non-canonical Wnt ligand, is overexpressed in high-grade GBM and GSCs being involved in GBM infiltration by regulating MMP-2 expression [26]. In addition, Binda et al. demonstrated that highly infiltrating mesenchymal glioblastoma cells were associated with the expression of Wnt5a and that its overexpression induces an invasive phenotype and activates the typical cell invasion genes in low-invading GSCs [27]. Furthermore, TGF-β signaling promotes GBM cells’ stemness through SOX4-SOX2 pathway activation, and Smad-dependent induction of LIF (leukemia inhibitory factor), which subsequently activates the Janus kinase (JAK)-signal transducer and activator of the transcription (STAT) pathway promoting the migration and invasion of GSCs [28,29][28][29]. TGF-β signaling regulates alpha V beta 3 integrin (vitronectin receptor), MMPs, and the tissue inhibitor of metalloproteinases (TIMP)-2 and cathepsin expression, which are relevant for ECM interaction and degradation during GSCs’ invasion [30]. The Hh signaling pathway is critical in GBM tumorigenesis, as well as in expressing transcription factor GLI1 [31]. Hh pathway activation promotes GSCs’ invasion and angiogenesis, thereby enhancing snail, slug, and vascular endothelial growth factor (VEGF) expression [31,32][31][32]. Strong evidence suggests the importance of GSCs in the infiltrative nature of GBM. New therapeutic proposals can be designed to reduce the recurrence of patients diagnosed with brain tumors. Interestingly, it is not only the undifferentiated cells that affect the invasiveness of GBM, as cells that do not present malignant characteristics have a positive impact on the processes of migration and invasion of tumor cells (Figure 1).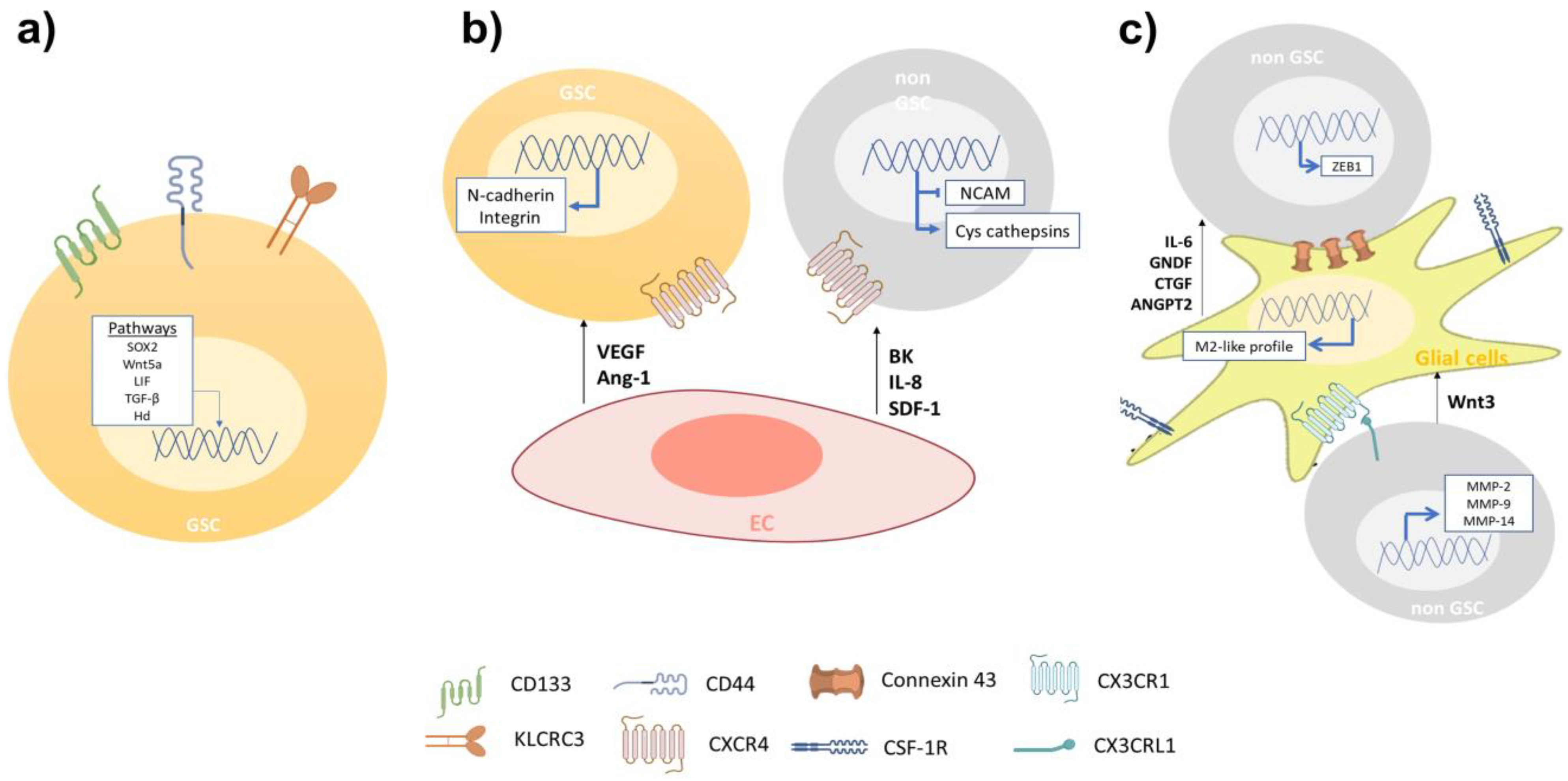
Figure 1. The migration and invasion of GBM cells is related to the inherent capacity and the cellular interactions that occur on the migratory front. (a) The invasiveness of GSCs is greater when it they are enriched in CD144, CD44, and KLRC3 markers. Along with the above, the activation of the signaling pathways SOX2, Wnt5a, LIF, TGF-β, and Hh has been related to the invasive phenotype. (b) GBM cells infiltrate vascular areas and interact with ECs. These cells secrete factors such as VEGF, Ang-1, BK, IL-8, and SDF-1, which promote cell invasion by expressing adhesion proteins such as N-cadherin and integrin on GSCs and non-GSCs. (c) Glial cells secrete IL-6, GNDF, CTGF, and ANGPT2, which promote the invasion of GBM cells (not GSC). On the other hand, connexin 43, whose expression is elevated in astrocytes associated with gliomas, is an important gap junction protein that allows direct communication between astrocytes and GBM cells. Activation of CX3CL1/CX3CR1 in tumor-associated microglia/macrophages (TAMs) increases the adhesion/migration capacity of GBM cells through the expression of MMP-2, -9, and -14.Finally, the secretion of Wnt3 by non-GSCs towards microglial cells, induce the transition to profile like M2; this activation increases the invasion of GBM cells.
3.1.2. Endothelial Cells
Inside the tumor tissue, GSCs are in contact with endothelial cells (ECs), creating a perivascular niche, which impacts the GBM’s progression. The perivascular niche is identified with the presence of the molecular markers such as CD31, CD34, CD133, nestin, α-SMA (alpha smooth muscle actin), GFAP (glial fibrillary acidic protein), CD14, and CD44 [33]. GSCs’ infiltration can be promoted via VEGF secreted by ECs, which may induce the transdifferentiation of GSCs into ECs, promoting angiogenesis and invasiveness [34]. The recently developed on-chip platform has further permitted live-cell imaging of GSC–microvessel interaction, enabling quantitative analysis of GSC polarization and migration, showing the importance of VEGF released from ECs for the invasiveness of GSCs [35]. Other soluble factors, such as angiopoietin (Ang)-1, regulate the crosstalk between glioma cells and ECs. The signaling axis between tyrosine kinase receptor Tie2/TEK and Ang-1 promotes cell invasion by regulating the expression of adhesion proteins, such as N-cadherin and integrin β1 [36]. ECs have been reported to attract GBM cells to blood vessels through the release of bradykinin (BK), which can activate the signal transduction pathways for B1 and B2 bradykinin receptors present in glioma cells [37]. Montana et al. have demonstrated that BK contributes to cell migration by activating Ca2+ intracellular mobilization. B2 receptor knockdown has shown that the association between blood vessels and GBM cells is decreased in rat brain slices [37]. Yadav et al. indicate that the overexpression of C-X-C chemokine receptor type 4 (CXCR-4), also known as fusin or CD184, in primary cultures of human GSCs and mouse glioma cells exhibits significant migration towards human (HBMVE) and mouse (MBMVE) brain microvascular ECs [38]. CXCL12 appears to be the main factor regulating cell migration when cells are attracted by MBVE and HBMVE. Additionally, the knockdown of CXCR-4 in mouse glioma cells inhibits the in vitro migration towards MBVE cells, decreasing perivascular invasion [38]. Additionally, the intracranial inoculation of CXCR-4 knockdown glioma cells reduced tumor growth and perivascular invasion, resulting in more sensitivity to radiotherapy, leading to increased survival [38]. McCoy et al. reported in a co-culture of patient-derived GBM cells and ECs an increased GBM invasiveness due to ECs via interleukin (IL)-8. In addition, the intracranial co-injection of GBM with ECs in an orthotopic mice model increased the tumor volume and led to less localized and more widely spread tumor formation. Both the in vitro and in vivo blockage of the IL-8 signaling reversed the GBM growth and cell invasion induced by ECs [39]. In a different study, ECs (HMEC-1) secreted stromal cell-derived factor-1 (SDF-1, also known as CXCL12), increasing the expression of cysteine cathepsins (B and S), MMP-9, and downregulating the endogenous cell adhesion molecule NCAM (neural cell adhesion molecule, also called CD56), in this way enhancing the invasiveness in U87MG cells [40]. The SDF-1 neutralizing antibody blocked endothelial cell-enhanced invasion of U87 cells through the downregulation of MMP-9, which suggests that under normoxic conditions GBM cells may be attracted by ECs [40]. All together, these data indicate that ECs-related molecules are potential therapeutic targets to impair perivascular GBM invasion (Figure 1).3.1.3. Glial Cells
The GBM TME includes different non-cancer cells, such as astrocytes, oligodendrocytes, and microglia, that are able to support tumor growth [41]. Astrocytes constitute about 50% of the human brain volume, having an important role in brain physiology and diseases [42]. The GBM cells can activate surrounding astrocytes (astrogliosis), which secrete high amounts of chemokines, such as IL-6, thereby enhancing GBM cell invasion and tissue infiltration by increasing the expression of MMPs [43]. In addition, GNDF (glial cell line-derived neurotrophic factor), secreted by astrocytes, induces the invasion of GBM cells by activating RET (rearranged during transfection)/GFRα1 (GDNF family receptor alpha-1) receptors and pro-tumoral signaling pathways, such as MAPK (mitogen-activated protein kinases) and PI3K (phosphatidylinositol 3-kinase)/Akt [44]. Edwards et al. reported that reactive astrocytes secrete CTGF (connective tissue growth factor), triggering NFkB (nuclear factor kappa B) signaling activation and the subsequent expression of ZEB1 in GBM cells, stimulating the epithelial–mesenchymal transition (EMT) and tumor cell infiltration [45]. This suggests that astrocytes are attracted to the GBM cells and facilitate their infiltration into the healthy brain tissue by the paracrine secretion of diverse molecules. Different studies describe that GBM cells are capable to release extracellular vesicles (EVs) into the tumor microenvironment. These tumor derived EVs are internalized by surrounding astrocytes, thus altering the expression and release of cytokines, which will promote tumor proliferation and invasion [46]. Furthermore, connexin 43 (Cx43), whose expression is elevated in glioma-associated astrocytes, is a major gap junction protein allowing direct communication between astrocytes and GBM cells [47]. Ectopic expression of a truncated Cx43 form altered the channel formation between GBM cells and astrocytes, decreasing GBM spreading into the brain parenchyma [48] (Figure 1). Oligodendrocytes also play important roles in brain physiology, by regulating neuronal activities, neural plasticity, and metabolic support [49]. Oligodendrocytes are localized in the border of the tumoral microenvironment and are able to stimulate GBM cell invasion [50]. Kawashima et al. reported that oligodendrocyte cells overexpress Ang-2, upregulating U251 and T98G cell lines invasiveness [51]. In addition, it has been reported that dual blockade of Ang-2/VEGF may prolong survival of GBM patients by reprogramming the tumor immune microenvironment and delaying tumor growth [52]. Tumor infiltrating immune cells account for nearly 30% of all tumor cells, including macrophages (T cells, NK cells, and macrophages derived from bone marrow) [53]. Different types of lymphocytes are capable of infiltrating into the tumor tissue, mainly CD4+ T helper, CD8+ T cytotoxic, and Tregs [54]; where CD4+ presents a higher number than CD8+, being associated with a higher degree of aggressiveness, these cellular types are called tumor-infiltrating lymphocytes (TILs) [55]. TILs have been associated with specific alterations to the transcript profile of GBM. Based on the analysis of 171 histopathological images obtained from the TCGA database, the authors find a relationship between lymphocyte infiltration and the histopathological and mutation characteristics of GBM. It was observed that mesenchymal subtype GBMs have a higher number of TILs, unlike proneural and classic subtypes. Additionally, these TILs were mainly associated with tumors with mutations in the NF1 and RB1 genes [56]. In addition, the release of pro-inflammatory cytokines released by T cells has been shown to contribute significantly to the invasiveness of GBM cells, suggesting that TILs play an important role in maintaining the characteristics of mesenchymal GBM [57]. Revi and collaborators, from a multidimensional study that integrates the gene expression of immune cells from 12 GBM samples, showed that there is a subset of TILs that secrete IL-10, specifically in regions of the GBM with mesenchymal characteristics, contributing to the maintenance of the invasive capacity as well as the immunosuppressive microenvironment [58]. On the other hand, NK cells have been shown to play an important role in the negative regulation of GBM cell invasiveness. Studies in BALB/c-nude mice showed that inactivating NK cells led to an increase in widespread GBM metastasis [59]. GBM cells can inhibit the cytotoxic activity of NK cells by expressing MHC-I and/or PD-1, suggesting that these proteins may play an important role in promoting the invasion of GBM cells into healthy tissue [60,61][60][61]. The authors suggest NK cells play important inhibitory role over extracranial metastasis, this is possibly through direct interaction between GBM cells and NK cells, in areas where the immune cells monitor the abnormal cells [59]. Microglia are resident myeloid cells in the central nervous system (CNS) that control homeostasis and protect the CNS from damage and infections. Microglia and peripheral myeloid cells accumulate and adapt tumor-supporting invasiveness of GBM cells, through the release of several chemoattractants [62]. The activation of CX3CL1 (chemokine C-X3-C motif ligand 1, also known as fractalkine)/CX3CR1 (CX3C chemokine receptor 1) in tumor-associated microglia/macrophages (TAMs) increases the adhesion/migration capacity of GBM cells through the expression of MMP-2, -9, and -14 [63]. Markovic et al. studied the role of microglia in GBM invasion using brain slices ex vivo, and after removing the microglia with clodronate-filled liposomes, GBM cells were unable to infiltrate into healthy brain tissue, demonstrating the importance of this cell type in GBM invasion [64]. Furthermore, it has been reported that the microglia-GBM cells’ crosstalk modulates tumor infiltration by the activation of epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling [65]. Ye et al. reported GSCs CD133(+) became more aggressive after being co-cultured with TAMs; however, the GSCs’ invasion was inhibited by the neutralization of TGF-β1, decreasing the expression of MMP-9 in GBM cells [66]. Moreover, Wnt signaling is one of the main pathways related to tumor progression and cell invasion [67]. Wnt3a released by GBM cells can stimulate the activation of Wnt/β-catenin pathways in microglia cells, thereby inducing a protumor M2-like profile in these cells; this activation increases the invasion of the GBM cells [68]. Kloepper et al. reported using mice bearing orthotopic syngeneic (Gl261) GBM and human (MGG8) GBM xenografts and found that the dual blockade of Ang-2/VEGF reprogrammed the protumor M2 macrophages toward the antitumor M1 phenotype, improving the survival of GBM mice [69]. Historically, the activation of TAMs has been classified as the proinflammatory M1 and immunosuppressive M2 state; however, the use of single cell RNA sequencing (scRNA-seq) has allowed the identification of new molecular patterns in this cell type. One scRNA-seq study from human biopsies identified that CD11b+ TAMs were capable of simultaneously expressing M1 and M2 activation markers [70]. On the other hand, a study using flow cytometry and scRNAseq revealed that CD11b+ cells derived from xenografted tumors present a remarkable functional heterogeneity, in which the subpopulation of TAMs expressing Ccl22, Cd274 (encoding PD-L1), and Ccl5 supports a functional immunosuppressive state [71]. Another study based on scRNA-seq analysis of 20.1986 cells, in which are included glioma, immune, and stromal cells reported that immune cells infiltrating tumor tissue show extensive molecular heterogeneity, identifying nine myeloid subtypes, indicating that S100A4 expression is a regulator of immunosuppressive T and myeloid cells in GBM [72] Cui et al. using scRNAseq data analysis covering a total of 16.201 GBM-infiltrating immune cells, demonstrated the existence of new microglia subgroups (primed and repressed), which this subgroup varied according to GBM subtype [73], where the proneural subtype was characterized by primed microglia, and the classical repressed microglia [73]. Unfortunately, of the nine GBM tissues analyzed from which data were obtained, GBMs of the mesenchymal subtype were not included; however, due to the high intracellular heterogeneity of this malignant tumor, it has been shown that regions of GBM exhibit characteristics of the different subtypes, suggesting that there is a distinct immune cell infiltration [74]. Experimental evidence published by different research groups shows that the analysis of single cells in GBM is opening the possibility of identifying and understanding the plasticity of TAMs, clearly indicating the existence of new states in addition to M1 and M2, as initially understood.3.2. Non-Cellular Components
Extracellular Matrix
The ECM comprises ~20% of the brain mass [75] and its function is not only to provide structural and biochemical support to surrounding cells, but to also play a key role in the regulation of several cellular processes, such as brain tissue homeostasis, viability, cell differentiation, migration, and invasion [75]. The brain’s ECM major components are hyaluronic acid (HA), tenascin-C (TNC), laminin, and collagen, among others, which have a key role in modulating invasiveness [76,77][76][77]. Infiltrative tumor cells are capable of remodeling and degrading ECM by MMPs released into the extracellular space [78]. MMP-2 and MMP-9 are the most highly expressed MMPs in GBM tissue, and they have been linked to a poor patient prognosis [79]. GBM cells are able to modify ECM components, promoting infiltration. Wiranowska et al. demonstrated in human U373 and mouse G26 GBM cell lines that the activation of CD44 by its ligand HA stimulates the synthesis and secretion of HA [80]. Additionally, Annabi et al. reported that CD44/HA interaction promotes glioma cell infiltration into healthy brain tissue by the upregulation of MT1-MMP expression [81]. Osteopontin (OPN), another component of the ECM, has been identified as a CD44 ligand, so its expression has been proposed as a possible modulator of GBM aggressiveness [82]. CD44/OPN have a perivascular expression pattern and their interaction induces the activation of the γ-secretase-regulated intracellular domain of CD44, which promotes migration, invasion, and stem cell-like phenotypes of GBM cells via CBP (CREB binding protein)/p300-dependent enhancement of HIF (hypoxia-inducible factor)-2α activity [82]. Furthermore, the co-expression of CD44 and OPN has been identified in GBM perivascular tissues and has been related to the stem-like phenotype [82]. Another ECM component is TNC glycoprotein, which has been implicated in embryogenesis, wound healing, and tumor progression [83]. It has been reported that elevated levels of TNC are correlated with GBM patients’ poor prognosis [84]. GBM cells produce and release TNC into the extracellular space, thereby changing the composition of the surrounding tissue, which facilitates the cellular invasion process [85]. Hirata et al. demonstrated that TNC silencing does not affect the proliferation of GBM cells, but impairs the in vitro cell migration in a two-dimensional substrate and decreases the cellular infiltration towards the brain parenchyma in a mouse xenograft model [86]. On the other hand, it has been reported that the downregulation of TNC in the TME causes less tumor cell infiltration but increases proliferation and the growth of the tumor mass. These results suggest that TNC could act as an important modulator of the so-called “go or grow” phenomenon [87,88][87][88]. This hypothesis proposes that adherent cells reversibly switch between migratory and proliferative phenotypes, where cells in the migratory state are more mobile than those in the proliferative state, because they are not using energy for proliferation [88]. Another study showed that MMP-12 is upregulated in GBM cells when they are exposed to a three-dimensional matrix enriched in TNC, and its silencing decreases in vitro cell invasion through MMP-12 downregulation [89]. Additionally, TNC could stimulate GSCs invasiveness by ADAM9 metalloproteinase expression and activity, via the c-Jun NH2-terminal kinase pathway [90]. The ADAM9 relevance in the invasive phenotype of GSCs was evaluated in histological samples of GBM patients and orthotopic xenograft models, reporting an elevated co-expression of ADAM9 and TNC in the invasive front of the tumor tissue [90]. In a differential microarray expression analysis, in which healthy tissue was compared with GBM tumor tissue, Ljubimova et al. identified two genes that were constitutively expressed in GBM, EGFR and laminin-α4 [91]. The α4 chain of laminin is constituted by laminin-9, laminin-8, or laminin-14 [92]. They found that during the GBM tumor progression, a switch of laminin-9 to laminin-8 occurs in the α4 chain of the blood vessel basement membranes [93]. In fact, laminin-8 silencing impairs in vitro cell invasion by almost 50% [93]. Furthermore, the study of 37 glial primary tumors, including 23 GBMs, demonstrated that the overexpression of laminin-8 was strongly associated with reduced recurrence time after surgery and poor survival of GBM patients, suggesting a role of laminin-8 in GBM spreading and infiltration [94]. Another research group analyzed 57 GBM biopsies reporting high expression of laminin-2 and -5 in infiltrative areas, compared with the tumor core, suggesting that laminin is involved in GBM invasiveness [95]. Similarly like laminin, brevican (BCAN) is a proteoglycan exclusive to the central nervous system and its expression is increased in GBM compared to healthy brain tissue, and has been associated with aggressiveness at the late stage of glioma progression [96]. Mice intrathecal inoculation with human GSCs primary cultures demonstrated that BCAN was expressed mainly in GSC niches, promoting tumor cells’ infiltration [97]. Moreover, Nakada et al. demonstrated that ADAMTS-5, a member of the ADAMTS family, is overexpressed in GBM cells and degrades BCAN, promoting cell invasion [98] (Figure 2).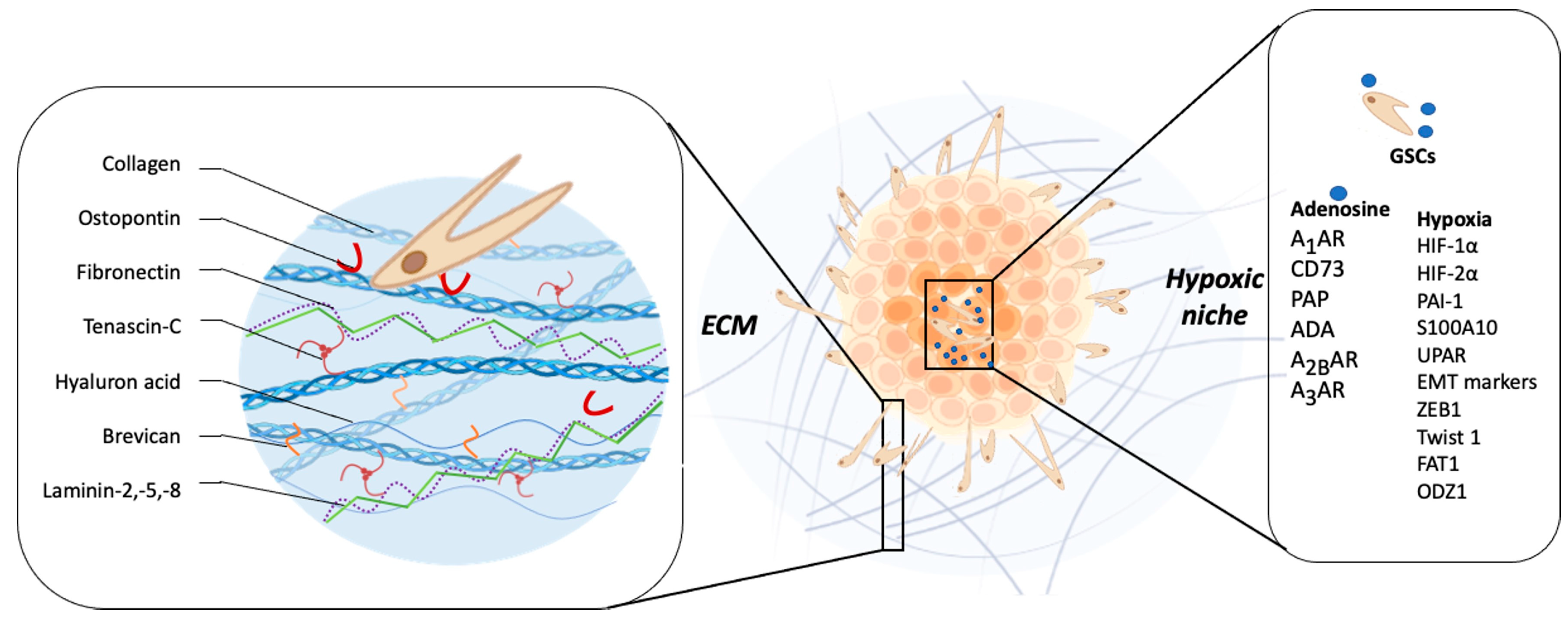
Figure 2. Relationship between tumor microenvironment components and GBM invasiveness. The main constituents of the brain ECM are collagen, osteopontin, fibronectin, tenascin-C, hyaluronic acid, brevican, laminin-2, -5, and -8, which have a role key in modulating invasiveness. The hypoxic microenvironment of GBM, characteristic of this tumor, promotes invasion mainly through the induction of one of the family of transcription factors induced under conditions of low O2 pressure (HIF-1 and 2), the increase in the expression of MMP-2 and MMP-9, plasminogen complex system (PAI-1), plasminogen receptor (S100A10), Upa receptor (uPAR), and markers of the epithelial–mesenchymal transition (EMT) process; expression and regulation of transcription factors such as ZEB1, Twist1, FAT1; as well as the induction of the epigenetic regulation of ODZ1. Adenosine has been recognized as one of the molecules that increases in the hypoxic niche of GBM. Proteins such as CD73, PAP, and ADA participate in its extracellular metabolism. This nucleoside regulates biological processes through the A1, A2B, and A3 adenosine receptors, ultimately promoting the invasive phenotype of GSCs and non-GSCs.
References
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312.
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8.
- Drappatz, J.; Norden, A.D.; Wen, P.Y. Therapeutic strategies for inhibiting invasion in glioblastoma. Expert Rev. Neurother. 2009, 9, 519–534.
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from in Vitro Models. Int. J. Mol. Sci. 2018, 19, 147.
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912.
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms Regulating Glioma Invasion. Cancer Lett. 2015, 362, 1–7.
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465.
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16.
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009.
- Cayre, M.; Canoll, P.; Goldman, J.E. Cell migration in the normal and pathological postnatal mammalian brain. Prog. Neurobiol. 2009, 88, 41–63.
- Mbeunkui, F.; Johann, D.J., Jr. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582.
- Wang, M.; Jingzhou, Z.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773.
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014.
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 15, 1203–1217.
- Ayuso-Sacido, A.; Moliterno, J.A.; Kratovac, S.; Kapoor, G.S.; O’Rourke, D.M.; Holland, E.C.; García-Verdugo, J.M.; Roy, N.S.; Boockvar, J.A. Activated EGFR signaling increases proliferation, survival, and migration and blocks neuronal differentiation in post-natal neural stem cells. Neurooncol. 2010, 97, 323–337.
- Yu, S.; Yang, X.; Zhang, B.; Ming, H.; Chen, C.; Ren, B.; Liu, Z.; Liu, B. Enhanced invasion in vitro and the distribution patterns in vivo of CD133+ glioma stem cells. Chin. Med. J. 2011, 124, 2599–2604.
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 23, 5387041.
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Ozaki, S.; Kanemura, Y.; Matsumoto, S.; Suehiro, S.; Ohtsuka, Y.; Kohno, S.; et al. Hypoxia-regulated expression of CD44 and osteopontin can change the phenotype of glioma stem-like cells from highly invasive to less invasive/proliferative tumors in glioblastoma. Transl. Oncol. 2021, 4, 101137.
- Cheray, M.; Bessette, B.; Lacroix, A.; Mélin, C.; Jawhari, S.; Pinet, S.; Deluche, E.; Clavère, P.; Durand, K.; Sanchez-Prieto, R.; et al. KLRC3, a Natural Killer receptor gene, is a key factor involved in glioblastoma tumourigenesis and aggressiveness. J. Cell. Mol. Med. 2017, 21, 244–253.
- Ortensi, B.; Setti, M.; Osti, D.; Pelicci, G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell. Res. Ther. 2013, 28, 18.
- Alonso, M.M.; Diez-Valle, R.; Manterola, L.; Rubio, A.; Liu, D.; Cortes-Santiago, N.; Urquiza, L.; Jauregi, P.; Lopez de Munain, A.; Sampron, N.; et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE 2011, 6, e26740.
- Tamase, A.; Muraguchi, T.; Naka, K.; Tanaka, S.; Kinoshita, M.; Hoshii, T.; Ohmura, M.; Shugo, H.; Ooshio, T.; Nakada, M.; et al. Identification of tumor-initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc. Natl. Acad. Sci. USA 2009, 106, 17163–17168.
- Mehta, S.; Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell. Mol. Life Sci. 2018, 75, 385–402.
- Arnés, M.; Casas Tintó, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222.
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53.
- Kamino, M.; Kishida, M.; Kibe, T.; Ikoma, K.; Iijima, M.; Hirano, H.; Tokudome, M.; Chen, L.; Koriyama, C.; Yamada, K.; et al. Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer Sci. 2011, 102, 540–548.
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-like Cells. Cancer Res. 2017, 77, 996–1007.
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.S.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009, 15, 315–327.
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955.
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185.
- Zhu, H.; Lo, H. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr. Genom. 2010, 11, 238–245.
- Maiti, S.; Mondal, S.; Satyavarapu, E.; Mandal, C. mTORC2 regulates hedgehog pathway activity by promoting stability to Gli2 protein and its nuclear translocation. Cell Death Dis. 2017, 8, e2926.
- Chen, J.; Mao, S.; Li, H.; Zheng, M.; Yi, L.; Lin, J.M.; Lin, Z.X. The pathological structure of the perivascular niche in different microvascular patterns of glioblastoma. PloS ONE 2017, 12, e0182183.
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280.
- Gerigk, M.; Bulstrode, H.; Shi, H.H.; Tönisen, F.; Cerutti, C.; Morrison, G.; Rowitch, D.; Huang, Y.Y.S. On-chip perivascular niche supporting stemness of patient-derived glioma cells in a serum-free, flowable culture. Lab Chip 2021, 21, 2343–2358.
- Liu, D.; Martin, V.; Fueyo, J.; Lee, O.H.; Xu, J.; Cortes-Santiago, N.; Alonso, M.M.; Aldape, K.; Colman, H.; Gomez-Manzano, C. Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget 2010, 1, 700–709.
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867.
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83837.
- Coy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.R.; Zipfel, W.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069.
- Kenig, S.; Alonso, M.B.; Mueller, M.M.; Lah, T.T. Glioblastoma and endothelial cells crosstalk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett. 2010, 1, 53–61.
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5.
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790.
- Chen, W.; Xia, T.; Wang, D.; Huang, B.; Zhao, P.; Wang, J.; Qu, X.; Li, X. Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 2016, 7, 62425–62438.
- Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Na’ara, S.; Gil, Z. Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int. J. Cancer 2015, 137, 1012–1020.
- Edwards, L.A.; Woolard, K.; Son, M.J.; Li, A.; Lee, J.; Ene, C.; Mantey, S.A.; Maric, D.; Song, H.; Belova, G.; et al. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion, n. J. Natl. Cancer Inst. 2011, 103, 1162–1178.
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581.
- Dong, H.; Zhou, X.W.M.; Wang, X.; Yang, Y.; Luo, J.W.; Liu, Y.H.; Mao, Q. Complex role of connexin 43 in astrocytic tumors and possible promotion of glioma-associated epileptic discharge (Review). Mol. Med. Rep. 2017, 16, 7890–7900.
- Sin, W.; Aftab, Q.; Bechberger, J.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516.
- Bercury, K.K.; Macklin, W.B. Dynamics and mechanisms of CNS myelination. Dev. Cell. 2015, 32, 447–458.
- Hide, T.; Shibahara, I.; Kumabe, T. Novel concept of the border niche: Glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell-like features. Brain Tumor Pathol. 2019, 36, 63–73.
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bähr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57.
- Kawashima, T.; Yashiro, M.; Kasashima, H.; Terakawa, Y.; Uda, T.; Nakajo, K.; Umaba, R.; Tanoue, Y.; Tamrakar, S.; Ohata, K. Oligodendrocytes Up-regulate the Invasive Activity of Glioblastoma Cells via the Angiopoietin-2 Signaling Pathway. Anticancer Res. 2019, 39, 577–584.
- Prionisti, I.; Bühler, L.H.; Walker, P.R.; Jolivet, R.B. Harnessing Microglia and Macrophages for the Treatment of Glioblastoma. Front. Pharmacol. 2019, 10, 506.
- Strepkos, D.; Markouli, M.; Klonou, A.; Piperi, C.; Papavassiliou, A.G. Insights in the immunobiology of glioblastoma. J. Mol. Med. 2020, 98, 1–10.
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Invest. 2017, 97, 498–518.
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960.
- Singh, K.; Hotchkiss, K.M.; Patel, K.K.; Wilkinson, D.S.; Mohan, A.A.; Cook, S.L.; Sampson, J.H. Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma. Cancers 2021, 13, 5367.
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kückelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13, 925.
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011.
- Xue, S.; Hu, M.; Li, P.; Ma, J.; Xie, L.; Teng, F.M.; Zhu, Y.; Fan, B.; Mu, D.; Yu, J. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget 2017, 30, 49702–49712.
- Burster, T.; Gärtner, F.; Bulach, C.M.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236.
- Gopinath, A.; Collins, A.; Khoshbouei, H.; Streit, W.J. Microglia and Other Myeloid Cells in Central Nervous System Health and Disease. J. Pharmacol. Exp. Ther. 2020, 375, 154–160.
- Held-Feindt, J.; Hattermann, K.; Müerköster, S.S.; Wedderkopp, H.; Knerlich-Lukoschus, F.; Ungefroren, H.; Mehdorn, H.M.; Mentlein, R. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell. Res. 2010, 316, 1553–1566.
- Markovic, D.S.; Glass, R.; Synowitz, M.; Van Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762.
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527.
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453.
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Iorio, P.D.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105.
- Matias, D.; Dubois, L.G.; Pontes, B.; Rosário, L.; Ferrer, V.P.; Balça-Silva, J.; Fonseca, A.C.C.; Macharia, L.W.; Romão, L.E.; Spohr, T.C.L.S.; et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/β-Catenin Signaling. Mol. Neurobiol. 2019, 56, 1517–1530.
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481.
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234.
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 19, 1151.
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 2022, 13, 767.
- Cui, X.; Wang, Q.; Zhou, J.; Wang, Y.; Xu, C.; Tong, F.; Wang, H.; Kang, C. Single-Cell Transcriptomics of Glioblastoma Reveals a Unique Tumor Microenvironment and Potential Immunotherapeutic Target Against Tumor-Associated Macrophage. Front. Oncol. 2021, 11, 710695.
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10, 494.
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729.
- Novak, U.; Kaye, A.H. Extracellular matrix, and the brain: Components and function. J. Clin. Neurosci. 2000, 7, 280–290.
- Ferrer, V.P.; Moura Neto, V.; Mentlein, R. Glioma infiltration and extracellular matrix: Key players and modulators. Glia 2018, 66, 1542–1565.
- Peng, W.J.; Yan, J.W.; Wan, Y.N.; Wang, B.X.; Tao, J.H.; Yang, G.J.; Pan, H.F.; Wang, J. Matrix metalloproteinases: A review of their structure and role in systemic sclerosis. J. Clin. Immunol. 2012, 32, 1409–1414.
- Hagemann, C.; Anacker, J.; Ernestus, R.I.; Vince, G.H. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 2012, 3, 67–79.
- Wiranowska, M.; Ladd, S.; Moscinski, L.C.; Hill, B.; Haller, E.; Mikecz, K.; Plaas, A. Modulation of hyaluronan production by CD44 positive glioma cells. Int. J. Cancer 2010, 127, 532–542.
- Annabi, B.; Bouzeghrane, M.; Moumdjian, R.; Moghrabi, A.; Béliveau, R. Probing the infiltrating character of brain tumors: Inhibition of RhoA/ROK-mediated CD44 cell surface shedding from glioma cells by the green tea catechin EGCg. J. Neurochem. 2005, 94, 906–916.
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeckm, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369.
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adh. Migr. 2015, 9, 48–82.
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163.
- Behrem, S.; Zarković, K.; Eskinja, N.; Jonjić, N. Distribution pattern of tenascin-C in glioblastoma: Correlation with angiogenesis and tumor cell proliferation. Pathol. Oncol. Res. 2005, 11, 229–235.
- Hirata, E.; Arakawa, Y.; Shirahata, M.; Yamaguchi, M.; Kishi, Y.; Okada, T.; Takahashi, J.A.; Matsuda, M.; Hashimoto, N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009, 100, 1451–1459.
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517.
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65.
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780.
- Sarkar, S.; Zemp, F.J.; Senger, D.; Robbins, S.M.; Yong, V.W. ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor-initiating cells. Neuro Oncol. 2015, 17, 1095–1105.
- Ljubimova, J.Y.; Lakhter, A.J.; Loksh, A.; Yong, W.H.; Riedinger, M.S.; Miner, J.H.; Sorokin, L.M.; Ljubimov, A.V.; Black, K.L. Overexpression of alpha4 chain-containing laminins in human glial tumors identified by gene microarray analysis. Cancer Res. 2001, 15, 5601–5610.
- Miner, J.H.; Yurchenco, P.D. Laminin functions in tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 2004, 20, 255–284.
- Ljubimova, J.Y.; Fujita, M.; Khazenzon, N.M.; Ljubimov, A.V.; Black, K.L. Changes in laminin isoforms associated with brain tumor invasion and angiogenesis. Front. Biosci. 2006, 11, 81–88.
- Ljubimova, J.Y.; Fugita, M.; Khazenzon, N.M.; Das, A.; Pikul, B.B.; Newman, D.; Sekiguchi, K.; Sorokin, L.M.; Sasaki, T.; Black, K.L. Association between laminin-8 and glial tumor grade, recurrence, and patient survival. Cancer 2004, 101, 604–612.
- Guo, P.; Imanishi, Y.; Cackowski, F.C.; Jarzynka, M.J.; Tao, H.Q.; Nishikawa, R.; Hirose, T.; Hu, B.; Cheng, S.Y. Up-regulation of angiopoietin-2, matrix metalloprotease-2, membrane type 1 metalloprotease, and laminin 5 gamma 2 correlates with the invasiveness of human glioma. Am. J. Pathol. 2005, 166, 877–890.
- Zhang, H.; Kelly, G.; Zerillo, C.; Jaworski, D.M.; Hockfield, S. Expression of a cleaved brain-specific extracellular matrix protein mediates glioma cell invasion In vivo. J. Neurosci. 1998, 18, 2370–2376.
- Dwyer, C.A.; Bi, W.L.; Viapiano, M.S.; Matthews, R.T. Brevican knockdown reduces late-stage glioma tumor aggressiveness. J. Neurooncol. 2014, 120, 63–72.
- Nakada, M.; Miyamori, H.; Kita, D.; Takahashi, T.; Yamashita, J.; Sato, H.; Miura, R.; Yamaguchi, Y.; Okada, Y. Human glioblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol. 2005, 110, 239–246.
- Ohnishi, T.; Hiraga, S.; Izumoto, S.; Matsumura, H.; Kanemura, Y.; Arita, N.; Hayakawa, T. Role of fibronectin-stimulated tumor cell migration in glioma invasion in vivo: Clinical significance of fibronectin and fibronectin receptor expressed in human glioma tissues. Clin. Exp. Metastasis 1998, 16, 729–741.
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462.
- Yu, Q.; Xue, Y.; Liu, J.; Xi, Z.; Li, Z.; Liu, Y. Fibronectin Promotes the Malignancy of Glioma Stem-Like Cells Via Modulation of Cell Adhesion, Differentiation, Proliferation and Chemoresistance. Front. Mol. Neurosci. 2018, 11, 130.
- Vidal, V.; Gutierrez, O.; Talamillo, A.; Velasquez, C.; Fernandez-Luna, J.L. Glioblastoma invasion factor ODZ1 is induced by microenvironmental signals through activation of a Stat3-dependent transcriptional pathway. Sci. Rep. 2021, 11, 16196.
More