Hashimoto’s thyroiditis (HT) is a gender autoimmune disease that is manifested by chronic inflammation of the thyroid. Clinical trial studies (CTSs) use molecular biotechnologies (MB) to approach HT appearance.
- Hashimoto’s thyroiditis
- molecular biotechnologies
1. Introduction
1.1. HT Biomarkers and Epidemiological Data
Overall, HT diffusion can be reported by considering two distinct types of serum biomarkers. Firstly, HT epidemiological information can be compiled based on autoimmune biomarkers of thyroid inflammation. Secondly, to archive HT epidemiological data, HT diffusion can be related to biochemical markers of hy-T and then to the onset of hy-T symptoms. By focusing on serum immunological biomarkers, HT is considered a gender-functional disorder [26]. This is because of the mechanisms underlying the appearance of autoantibodies [27,28][27][28]. In fact, the disruption of immune tolerance is genetically driven [29,30][29][30]. Particularly, HT autoimmune anomalies are genetically based on gender and pre-existing individual susceptibility [27,29,30][27][29][30]. In turn, the environment plays a critical role in altering genetic backgrounds by influencing disease development [26,30][26][30]. Hence, HT is reported in women 10–15 times more often than in men, with an incidence peak at around 30–50 years of age [31]. Conversely, in men, the HT incidence increases with aging and the incidence peak is reached 10–15 years later [31]. When HT diffusion is related to hy-T incidence, substantial differences emerge between hy-T that spreads in endemic areas of iodine deficiency and that which is accompanied by HT. Zimmermann and colleagues previously observed that hy-T typically emerges in HT patients independently according to iodine nutrition status [32,33][32][33]. This is because hy-T develops even in HT patients living in areas with sufficient iodine intake. Moreover, when populations are resident in iodine-deficient localities, it is quite common to find endemic hy-T [34]. Gender differences come up even when HT is related to the onset of hy-T signs and symptoms. In fact, distinctive clinical courses and different outcomes are observed in women compared to men. Further, differences in gender are a key determinant even of therapeutic responses to L-T4 [26,35][26][35]. Usually, hy-T symptoms include fatigue, cold intolerance, and constipation. However, there is a large variation in the clinical presentation of symptoms [15]. In women, hy-T develops more frequently at a later age than HT, especially after 60 years of age [26]. In addition, hy-T symptoms have no determinant role in the identification of endocrine disorders. This is because hy-T symptoms may occur in healthy female subjects, too. Lastly, L-T4 therapy may be associated with residual symptoms despite normal thyroid tests [17,24][17][24]. In men, hy-T symptoms that accompany overt HT are more recurrent, last longer, and are usually less treatable [26]. Therefore, the presence or absence of symptoms may be contributing factors in the identification of hy-T. Lastly, L-T4 therapy is less frequently accompanied by other side effects in men.1.2. Prevalence of HT Diagnoses
The methods used to diagnose HT have a long history related to the description of morphological alterations of the thyroid gland, the recognition of autoimmune pathogenesis, and the identification of thyroid hormones [4]. For a proper diagnosis of HT, several methods are involved; further, different biomarkers are assessed independently or in combination with each other [4]. Mainly, serum, ultrasound, and pathological examinations are considered HT diagnostic methods [4,31][4][31]. Current research has reported the global prevalence of diagnoses of HT according to different diagnostic methods [31]. Moreover, data about the prevalence of methods useful to confirm HT diagnoses have been provided [31]. Therefore, HT is prevalently diagnosed by ultrasonography (13.2%) and pathological examination (12.5%) [31]. When the serum autoantibody profile is considered, the prevalence rate of HT diagnosis stands at 7.8% (see Figure 9 in Ref. [31]) [31]. The combination of two methods, including serum antibody titers and color Doppler ultrasound, is used for HT diagnosed with a prevalence of 10.4% [31]. This prevalence is considerably lower (4.7%) if three methods, such as autoantibody titers, color Doppler ultrasonography, and fine needle aspiration, are combined. To confirm HT diagnosis, thyroid tissue alone is prevalently used (14.1%) [31].1.3. Molecular Biotechnologies (MB) and HT
MB are pivotal for new biomedical methodologies because of their capability to reveal molecular pathogenetic pathways as well as the genetic susceptibility of populations to develop AIDT [6]. Above all, now the use of genetic analysis is turning out to be a key tool for clinical genomic investigations owing to its high accuracy, reproducibility, and reliability of results. The effective clinical application of MB can be assessed in accordance with the advice of qualified clinical trial studies (CTSs) [36]. These investigations are the basis of genomic screenings designed to detect viral genetic material involved in the pathogenesis of diseases. Not only this, but CTSs can especially test how well genomic screenings work to identify susceptibility to develop diseases in subgroups of populations belonging to a specific continent. Molecular alterations occurring in the context of HT play crucial roles in promoting the cellular proliferation of both lymphocytes and glandular tissue. Indeed, mucosa-associated lymphoid tissue (MALT) lymphomas can originate at the site of HT [37,38][37][38]. On the other hand, for a considerable time, HT was reported concurrent with cancerous follicular lesions such as nodular goiter, adenoma, and carcinoma [38,39,40,41][38][39][40][41]. MB are currently employed for the classification of MALT lymphoma [42,43][42][43]. Further, these analyses are applied in dubious diagnoses of thyroid glandular cancerous lesions [44]. Mainly, these are part of innovative thyroid medicine that aims through biomarkers to early molecular diagnosis, personalized treatment, the prediction of cancerous risk, and prognostic information [45].2. Medical Applications of Molecular Biotechnologies in Hashimoto’s Thyroiditis
HT may appear through different clinical and histological aspects, and thus, morphological and serum diagnoses of HT are not equivalent [4]. In addition, HT may be associated with benign and malignant follicular lesions as well as lymphomatous proliferations [38,41,42][38][41][42]. The exact etiology of HT still remains incompletely elucidated. Mainly, it has been related to interactions of different elements, such as genetic alterations, environmental and epigenetic factors [30,47,48][30][46][47]. MB are promising surveying methods to apply to the HT population.
Viral and bacterial infections are currently involved in HT pathogenesis via multiple and often intertwined pathways. Based on the old Th1/Th2 paradigm, the so-called hygiene hypothesis (HH) has been adapted to the infective etiology of AIDT at the end of the last century [49,50,51,52][48][49][50][51]. Briefly, this hypothesis postulates that early infections in childhood protect against the establishment of autoimmunity [49,52,53,54,55][48][51][52][53][54]. Further, reduced exposure to microbial environments in childhood is considered an element conducive to the increase of autoimmune diseases in adults [56][55]. This is because an immune system educated by pathogen exposition may better suppress autoimmunity. However, the extension of HH to support HT pathogenesis has not reported a complete agreement [52][51]. Closely related to HH are the socio-demographic profiles of the HT population, data from migration surveys and biographic info of HT patients. By different concentrations, HT subjects are geographically distributed on the continental territories. A geographical map created on the bases of demographic observations reveals higher concentrations of HT subjects in Africa and Oceania (Figure 1) [31]. On the basis of socio-demographic observations, two divergent findings have been recorded. In low- and middle-income countries, the highest prevalence of HT patients is found among low-middle-income subjects (11.4%; see Figure 8 in Ref. [31]) [31]. However, HT patients are prevalently concentrated in high-income countries [31]. Therefore, the HH pathogenetic concepts can be applied to the last phenomena, whereas the first evidence seems limited only to the infectious etiology of HT. For over 50 years, surveys on the transmigration of populations are persistently reporting that subjects migrating from a country with a low incidence of autoimmune disorders develop immune-related diseases with the same frequency as the original inhabitants of the host country [53,57,58,59,60,61,62][52][56][57][58][59][60][61]. These data suggest an environmental effect at the beginning of autoimmune diseases. By reporting the biographic info of HT patients, several investigations have focused on the surprising association occurring between the birth month of individuals and HT. Mostly, HT patients were born in winter and autumn [63][62]. This data suggests that cold weather protects against TPO-Ab development [64][63]. Nevertheless, this evidence is consistent with the infective etiology of HT due to the abundant spread of infectious agents in winter. Further, these findings support HH because children born in winter have early exposure to infectious agents, facilitating the development of autoimmune diseases. However, moving from these premises, it is even possible to affirm that the incidence of HT for the individual subject may be predicted based on their date of birth. Summing up these phenomena, HH seems jarring with genetic features observed in autoimmune disorders, especially in HT. Molecular analyses have mapped on the short arm of chromosome 6 (6p) a super-region of 7.6 Mb, including the extended major histocompatibility complex (eMHC) [65,66][64][65]. This region lengthens telomerically from RPL12P1 to HIST1H2AA, and it is composed of six clusters and six super-clusters [66][65]. At 6p21.3 of eMHC, human leukocyte antigen (HLA) genes are localized, which are highly polymorphic [66][65]. HLA expressions are strongly related to infection, immunity, and inflammation [67][66]. In HT, genetic polymorphisms of HLA change depending on ethnicity [68][67]. This is because of different expressions of haplotypes in Caucasians (DR3, DR5, DQ7, DQB1*03, DQw7 or DRB1*04-DQB1*0301) with respect to Japanese (DRB4*0101, HLA-A2, DRw53) and Chinese (DRw9) HT patients [68][67]. Together, this data suggests that non-genetic factors trigger the onset of autoimmune disorders through an unidentified genetic background that is common to the entire HT population. Therefore, among phases composing HT pathogenesis, individual genetic susceptibility enters at a later stage than environmental factors. Genetic disparities in HLA profiles are established through the use of molecular techniques. These methods have the advantage of systematically arranging HLA haplotypes using symbols. The complexity of the nomenclature of HLA haplotypes has been organized using multiple molecular techniques [69][68]. The first molecular approach to displaying HLA alleles concerned the application of Sanger sequencing-based typing (PCR-SBT) methods [69][68]. High-throughput sequencing (HTS) methods, including next-generation “short-read” (NGS) and third-generation “long-read” sequencing methods, are the natural evolution of PCR-SBT. Lastly, Oxford Nanopore Technology MinION is progressively reorganizing the number of HLA alleles [70][69]. Genotyping investigations on Graves’s disease (GD) have identified novel HLA alleles through high-resolution NGS [71,72][70][71]. Further, methods based on machine learning are useful for predicting HLA subtypes in GD [73][72]. These investigations suggest matching different medical biotechnologies to better explain pathogenetic stages involving HLA haplotypes for the development of autoimmune disorders. By focusing on available molecular sources for CTSs, it appears that the parvo and polyoma viruses were investigated in the mCTSs. The role of viruses in inducing HT has been explored, but it is still not completely determined [52,74,75][51][73][74]. New data is becoming available regarding the roles of DNA and RNA viruses in triggering HT [76,77][75][76]. DNA viruses, namely, parvovirus 19 (B19V), human hepatitis C virus, and human herpes virus-6, have been associated with the viral pathogenesis of HT [76,77,78,79,80][75][76][77][78][79]. Among RNA viruses, human immunodeficiency virus (HIV) has been related to HT as it is able to activate the inflammatory immune response through IL-6 [81,82][80][81]. Particularly in HIV patients, this cytokine plays an important role by orchestrating the inflammatory cascade associated with HT [82][81]. The importance of IL-6 has been recognized even in animal models of DNA virus infection. In fact, IL-6 amounts are incremented in lung tissues of naïve Balb/c mice that received parvoviruses [83][82]. Parvoviruses are widespread in different countries on the American, European, and Asian continents [77][76]. Among DNA viruses, parvoviruses display the highest levels of replication and recombination [84][83]. These viruses can replicate autonomously or, conversely, recombine with a helper virus to be perpetuated [84][83]. The International Committee on Taxonomy of Viruses (ICTV) has reported members of the Parvoviridae family as small (~20 nm in diameter), icosahedral, non-enveloped viruses that have a small single-stranded DNA of 4–6 kb [85][84]. In 2020, the Executive Committee of the ICTV approved a revision for the taxonomy of the Parvoviridae family [86][85]. Although the definition to describe these viruses remained, genetic criteria used to demark members composing this family have been updated. The proposal criteria proceed from discoveries of new members of the Parvoviridae family through the application of HTS methods. Basically, the classification based on the association with the host has been abandoned because these viruses infect phylogenetically disparate hosts (see Table 1 in Ref. [86][85]) [86][85]. In this family, infectious agents for animals have been incorporated, showing a large host range. In fact, this is vast enough to include many phyla ranging from primates, mammals, and avian species to invertebrates [86][85]. Beyond this, the Parvoviridae family embraces pathogens for arthropod clades, namely the arachnids of the Chelicerata, that the molecular clock estimates go back to marine fossils of the late Cambrian period [87,88,89][86][87][88]. In 1975, Cossart and colleagues detected for the first-time B19V in serum samples of subjects screened for hepatitis B virus [90][89]. Thirty years later, Allander and colleagues discovered bocavirus 1 (HBoV1) in human samples of nasopharyngeal aspirates belonging to children with respiratory tract infections [91][90]. B19V1 may cause a widespread and self-limiting infection in children and adults, known as erythema infectiosum or fifth disease [92][91]. Both B19V and HBoV1 are pathogens for humans and have been detected in cancerous thyroid cells and HT lesions [76,93,94,95][75][92][93][94]. B19V and HBoV1 exhibit a particular tropism for the nuclear compartment. The host machinery for nuclear import of viral capsid is a critical step in the early phase of infection [96,97,98][95][96][97]. The capsid binding protein cleavage and polyadenylation specificity factor 6 plays a dominant role in directing integration to euchromatin of HBoV1 and lentivirus HIV-1, too [96,97,98][95][96][97]. During the later stages of infection, the replication of B19V leads to morphological changes in the nucleus. These are due to the spatial reorganization of chromatin that appears marginalized to the nuclear periphery by super-resolution microscopic examination [99][98].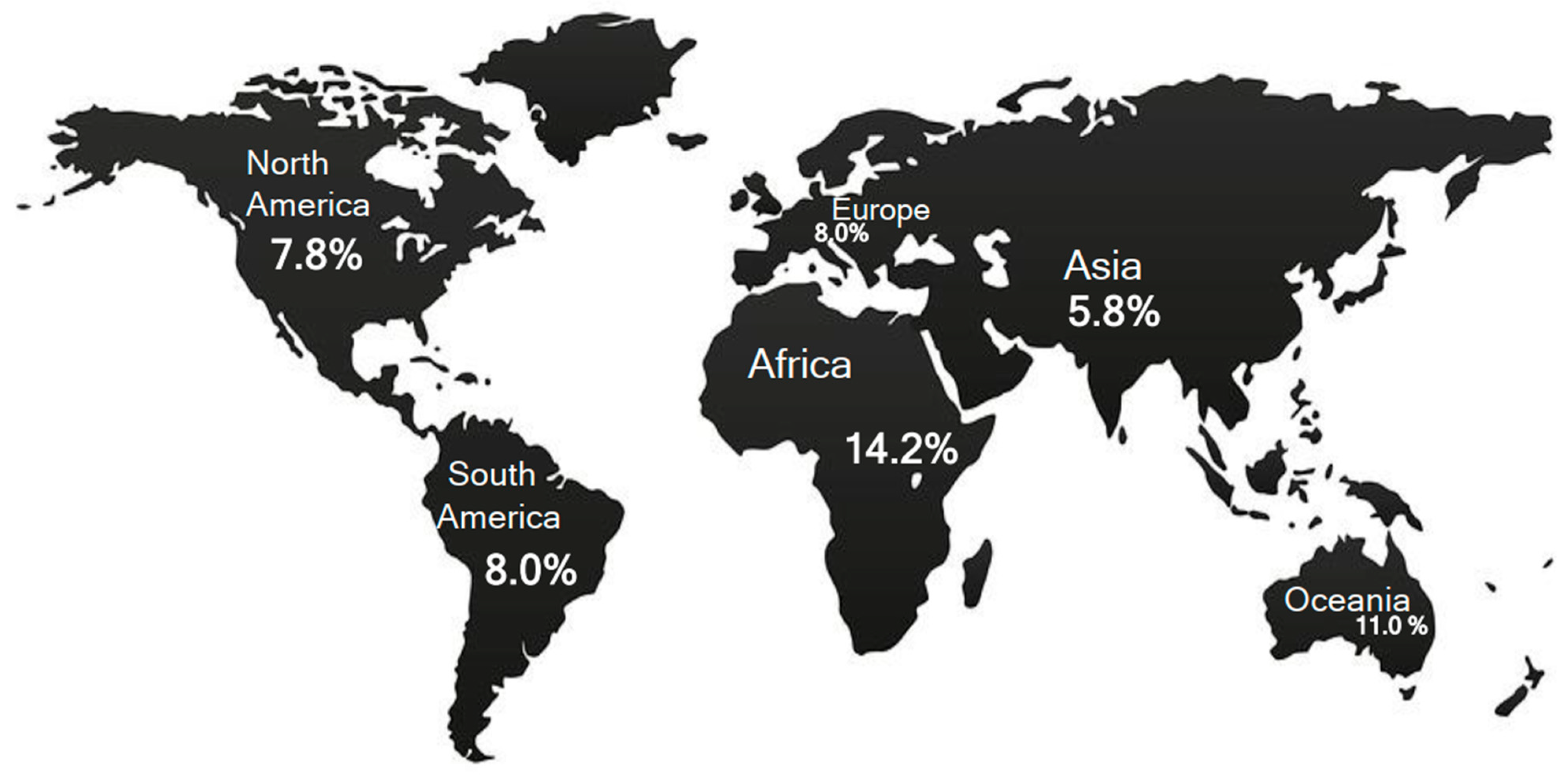
References
- Yoo, W.S.; Chung, H.K. Recent Advances in Autoimmune Thyroid Diseases. Endocrinol. Metab. 2016, 31, 379.
- Bliddal, S.; Nielsen, C.H.; Feldt-Rasmussen, U. Recent Advances in Understanding Autoimmune Thyroid Disease: The Tallest Tree in the Forest of Polyautoimmunity. F1000Research 2017, 6, 1776.
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune Thyroid Disorders. Autoimmun. Rev. 2015, 14, 174–180.
- Trovato, M. A Historical Excursus of Diagnostic Methods for Hashimoto Thyroiditis and Graves’ Disease. Gazz. Med. Ital. Arch. Sci. Med. 2020, 179, 479–485.
- Fröhlich, E.; Wahl, R. Thyroid Autoimmunity: Role of Anti-Thyroid Antibodies in Thyroid and Extra-Thyroidal Diseases. Front. Immunol. 2017, 8, 521.
- Bogusławska, J.; Godlewska, M.; Gajda, E.; Piekiełko-Witkowska, A. Cellular and Molecular Basis of Thyroid Autoimmunity. Eur. Thyroid J. 2022, 11, e210024.
- Thomas, T.; Sreedharan, S.; Khadilkar, U.N.; Deviprasad, D.; Kamath, M.P.; Bhojwani, K.M.; Alva, A. Clinical, Biochemical & Cytomorphologic Study on Hashimoto’s Thyroiditis. Indian J. Med. Res. 2014, 140, 729–735.
- Kahaly, G.J.; Gottwald-Hostalek, U. Use of Levothyroxine in the Management of Hypothyroidism: A Historical Perspective. Front. Endocrinol. 2022, 13, 1054983.
- Okosieme, O.; Gilbert, J.; Abraham, P.; Boelaert, K.; Dayan, C.; Gurnell, M.; Leese, G.; McCabe, C.; Perros, P.; Smith, V.; et al. Management of Primary Hypothyroidism: Statement by the British Thyroid Association Executive Committee. Clin. Endocrinol. 2016, 84, 799–808.
- Jordan, B.; Uer, O.; Buchholz, T.; Spens, A.; Zierz, S. Physical Fatigability and Muscle Pain in Patients with Hashimoto Thyroiditis. J. Neurol. 2021, 268, 2441–2449.
- Lei, Y.; Yang, J.; Li, H.; Zhong, H.; Wan, Q. Changes in Glucose-lipid Metabolism, Insulin Resistance, and Inflammatory Factors in Patients with Autoimmune Thyroid Disease. J. Clin. Lab. Anal. 2019, 33, e22929.
- Waliszewska-Prosół, M.; Bladowska, J.; Budrewicz, S.; Sąsiadek, M.; Dziadkowiak, E.; Ejma, M. The Evaluation of Hashimoto’s Thyroiditis with Event-Related Potentials and Magnetic Resonance Spectroscopy and Its Relation to Cognitive Function. Sci. Rep. 2021, 11, 2480.
- Jonklaas, J.; Bianco, A.C.; Bauer, A.J.; Burman, K.D.; Cappola, A.R.; Celi, F.S.; Cooper, D.S.; Kim, B.W.; Peeters, R.P.; Rosenthal, M.S.; et al. Guidelines for the Treatment of Hypothyroidism: Prepared by the American Thyroid Association Task Force on Thyroid Hormone Replacement. Thyroid 2014, 24, 1670–1751.
- Chaker, L.; Bianco, A.C.; Jonklaas, J.; Peeters, R.P. Hypothyroidism. Lancet 2017, 390, 1550–1562.
- Chaker, L.; Razvi, S.; Bensenor, I.M.; Azizi, F.; Pearce, E.N.; Peeters, R.P. Hypothyroidism. Nat. Rev. Dis. Prim. 2022, 8, 30.
- Razvi, S.; Korevaar, T.I.M.; Taylor, P. Trends, Determinants, and Associations of Treated Hypothyroidism in the United Kingdom, 2005–2014. Thyroid 2019, 29, 174–182.
- Wiersinga, W.M.; Duntas, L.; Fadeyev, V.; Nygaard, B.; Vanderpump, M.P.J. 2012 ETA Guidelines: The Use of L-T4 + L-T3 in the Treatment of Hypothyroidism. Eur. Thyroid J. 2012, 1, 55–71.
- Jonklaas, J.; Bianco, A.C.; Cappola, A.R.; Celi, F.S.; Fliers, E.; Heuer, H.; McAninch, E.A.; Moeller, L.C.; Nygaard, B.; Sawka, A.M.; et al. Evidence-Based Use of Levothyroxine/Liothyronine Combinations in Treating Hypothyroidism: A Consensus Document. Thyroid 2021, 31, 156–182.
- Lillevang-Johansen, M.; Abrahamsen, B.; Jørgensen, H.L.; Brix, T.H.; Hegedüs, L. Over- and Under-Treatment of Hypothyroidism Is Associated with Excess Mortality: A Register-Based Cohort Study. Thyroid 2018, 28, 566–574.
- Thayakaran, R.; Adderley, N.J.; Sainsbury, C.; Torlinska, B.; Boelaert, K.; Šumilo, D.; Price, M.; Thomas, G.N.; Toulis, K.A.; Nirantharakumar, K. Thyroid Replacement Therapy, Thyroid Stimulating Hormone Concentrations, and Long Term Health Outcomes in Patients with Hypothyroidism: Longitudinal Study. BMJ 2019, 366, l4892.
- Davies, T.F.; Morshed, S.A.; Mezei, M.; Latif, R. Brief Report—Monoclonal Antibodies Illustrate the Difficulties in Measuring Blocking TSH Receptor Antibodies. Front. Endocrinol. 2022, 13, 943459.
- Peterson, S.J.; Cappola, A.R.; Castro, M.R.; Dayan, C.M.; Farwell, A.P.; Hennessey, J.V.; Kopp, P.A.; Ross, D.S.; Samuels, M.H.; Sawka, A.M.; et al. An Online Survey of Hypothyroid Patients Demonstrates Prominent Dissatisfaction. Thyroid 2018, 28, 707–721.
- Perros, P.; Hegedüs, L.; Nagy, E.V.; Papini, E.; Hay, H.A.; Abad-Madroñero, J.; Tallett, A.J.; Bilas, M.; Lakwijk, P.; Poots, A.J. The Impact of Hypothyroidism on Satisfaction with Care and Treatment and Everyday Living: Results from E-Mode Patient Self-Assessment of Thyroid Therapy, a Cross-Sectional, International Online Patient Survey. Thyroid 2022, 32, 1158–1168.
- Bjerkreim, B.A.; Hammerstad, S.S.; Gulseth, H.L.; Berg, T.J.; Omdal, L.J.; Lee-Ødegård, S.; Eriksen, E.F. Effect of Liothyronine Treatment on Quality of Life in Female Hypothyroid Patients With Residual Symptoms on Levothyroxine Therapy: A Randomized Crossover Study. Front. Endocrinol. 2022, 13, 816566.
- Perros, P.; Nirantharakumar, K.; Hegedüs, L. Recent Evidence Sets Therapeutic Targets for Levothyroxine-Treated Patients with Primary Hypothyroidism Based on Risk of Death. Eur. J. Endocrinol. 2021, 184, C1–C3.
- Castello, R.; Caputo, M. Thyroid Diseases and Gender. Ital. J. Gend.-Specif. Med. 2019, 5, 136–141.
- Matana, A.; Popović, M.; Boutin, T.; Torlak, V.; Brdar, D.; Gunjača, I.; Kolčić, I.; Boraska Perica, V.; Punda, A.; Polašek, O.; et al. Genome-Wide Meta-Analysis Identifies Novel Gender Specific Loci Associated with Thyroid Antibodies Level in Croatians. Genomics 2019, 111, 737–743.
- Ragusa, F.; Fallahi, P.; Elia, G.; Gonnella, D.; Paparo, S.R.; Giusti, C.; Churilov, L.P.; Ferrari, S.M.; Antonelli, A. Hashimotos’ Thyroiditis: Epidemiology, Pathogenesis, Clinic and Therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101367.
- Pyzik, A.; Grywalska, E.; Matyjaszek-Matuszek, B.; Roliński, J. Immune Disorders in Hashimoto’s Thyroiditis: What Do We Know So Far? J. Immunol. Res. 2015, 2015, 979167.
- Vargas-Uricoechea, H. Molecular Mechanisms in Autoimmune Thyroid Disease. Cells 2023, 12, 918.
- Hu, X.; Chen, Y.; Shen, Y.; Tian, R.; Sheng, Y.; Que, H. Global Prevalence and Epidemiological Trends of Hashimoto’s Thyroiditis in Adults: A Systematic Review and Meta-Analysis. Front. Public Health 2022, 10, 1020709.
- Zimmermann, M.B.; Jooste, P.L.; Pandav, C.S. Iodine-Deficiency Disorders. Lancet 2008, 372, 1251–1262.
- Andersson, M.; Karumbunathan, V.; Zimmermann, M.B. Global Iodine Status in 2011 and Trends over the Past Decade. J. Nutr. 2012, 142, 744–750.
- Zimmermann, M.B. Iodine Deficiency. Endocr. Rev. 2009, 30, 376–408.
- Devdhar, M.; Drooger, R.; Pehlivanova, M.; Singh, G.; Jonklaas, J. Levothyroxine Replacement Doses Are Affected by Gender and Weight, But Not Age. Thyroid 2011, 21, 821–827.
- Poste, G.; Carbone, D.P.; Parkinson, D.R.; Verweij, J.; Hewitt, S.M.; Jessup, J.M. Leveling the Playing Field: Bringing Development of Biomarkers and Molecular Diagnostics up to the Standards for Drug Development. Clin. Cancer Res. 2012, 18, 1515–1523.
- Troch, M.; Woehrer, S.; Streubel, B.; Weissel, M.; Hoffmann, M.; Müllauer, L.; Chott, A.; Raderer, M. Chronic Autoimmune Thyroiditis (Hashimoto’s Thyroiditis) in Patients with MALT Lymphoma. Ann. Oncol. 2008, 19, 1336–1339.
- Trovato, M.; Giuffrida, G.; Seminara, A.; Fogliani, S.; Cavallari, V.; Ruggeri, R.M.; Campennì, A. Coexistence of Diffuse Large B-Cell Lymphoma and Papillary Thyroid Carcinoma in a Patient Affected by Hashimoto’s Thyroiditis. Arch. Endocrinol. Metab. 2017, 61, 643–646.
- Anil, C.; Goksel, S.; Gursoy, A. Hashimoto’s Thyroiditis Is Not Associated with Increased Risk of Thyroid Cancer in Patients with Thyroid Nodules: A Single-Center Prospective Study. Thyroid 2010, 20, 601–606.
- Chen, Y.-K.; Lin, C.-L.; Cheng, F.T.-F.; Sung, F.-C.; Kao, C.-H. Cancer Risk in Patients with Hashimoto’s Thyroiditis: A Nationwide Cohort Study. Br. J. Cancer 2013, 109, 2496–2501.
- Resende De Paiva, C.; Grønhøj, C.; Feldt-Rasmussen, U.; Von Buchwald, C. Association between Hashimoto’s Thyroiditis and Thyroid Cancer in 64,628 Patients. Front. Oncol. 2017, 7, 53.
- Pavlidis, E.T.; Pavlidis, T.E. A Review of Primary Thyroid Lymphoma: Molecular Factors, Diagnosis and Management. J. Investig. Surg. 2019, 32, 137–142.
- Rodríguez-Sevilla, J.J.; Salar, A. Recent Advances in the Genetic of MALT Lymphomas. Cancers 2021, 14, 176.
- Rossi, E.D.; Vielh, P. Thyroid and Molecular Testing. Advances in Thyroid Molecular Cytopathology. J. Mol. Pathol. 2021, 2, 77–92.
- Trovato, M. Update on International Medical Taxonomies of Biomarkers and Their Applications in Management of Thyroid Cancers. Diagnostics 2022, 12, 662.
- Lee, H.J.; Stefan–Lifshitz, M.; Li, C.W.; Tomer, Y. Genetics and Epigenetics of Autoimmune Thyroid Diseases: Translational Implications. Best Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101661.
- Ralli, M.; Angeletti, D.; Fiore, M.; D’Aguanno, V.; Lambiase, A.; Artico, M.; De Vincentiis, M.; Greco, A. Hashimoto’s Thyroiditis: An Update on Pathogenic Mechanisms, Diagnostic Protocols, Therapeutic Strategies, and Potential Malignant Transformation. Autoimmun. Rev. 2020, 19, 102649.
- Strachan, D.P. Hay Fever, Hygiene, and Household Size. BMJ 1989, 299, 1259–1260.
- Tomer, Y.; Davies, T.F. Infection, Thyroid Disease, and Autoimmunity. Endocr. Rev. 1993, 14, 107–120.
- Desailloud, R.; Hober, D. Viruses and Thyroiditis: An Update. Virol. J. 2009, 6, 5.
- Morohoshi, K.; Takahashi, Y.; Mori, K. Viral Infection and Innate Pattern Recognition Receptors in Induction of Hashimoto’s Thyroiditis. Discov. Med. 2011, 12, 505–511.
- Okada, H.; Kuhn, C.; Feillet, H.; Bach, J.-F. The ‘Hygiene Hypothesis’ for Autoimmune and Allergic Diseases: An Update. Clin. Exp. Immunol. 2010, 160, 1–9.
- Versini, M.; Jeandel, P.-Y.; Bashi, T.; Bizzaro, G.; Blank, M.; Shoenfeld, Y. Unraveling the Hygiene Hypothesis of Helminthes and Autoimmunity: Origins, Pathophysiology, and Clinical Applications. BMC Med. 2015, 13, 81.
- Garn, H.; Potaczek, D.P.; Pfefferle, P.I. The Hygiene Hypothesis and New Perspectives—Current Challenges Meeting an Old Postulate. Front. Immunol. 2021, 12, 637087.
- Kondrashova, A.; Seiskari, T.; Ilonen, J.; Knip, M.; Hyöty, H. The ‘Hygiene Hypothesis’ and the Sharp Gradient in the Incidence of Autoimmune and Allergic Diseases between Russian Karelia and Finland. APMIS 2013, 121, 478–493.
- Detels, R.; Brody, J.A.; Edgar, A.H. Multiple Sclerosis among American, Japanese and Chinese Migrants to California and Washington. J. Chronic Dis. 1972, 25, 3–10.
- Leibowitz, U.; Kahana, E.; Alter, M. The Changing Frequency of Multiple Sclerosis in Israel. Arch. Neurol. 1973, 29, 107–110.
- Bodansky, H.J.; Staines, A.; Stephenson, C.; Haigh, D.; Cartwright, R. Evidence for an Environmental Effect in the Aetiology of Insulin Dependent Diabetes in a Transmigratory Population. BMJ 1992, 304, 1020–1022.
- Staines, A.; Hanif, S.; Ahmed, S.; McKinney, P.A.; Shera, S.; Bodansky, H.J. Incidence of Insulin Dependent Diabetes Mellitus in Karachi, Pakistan. Arch. Dis. Child. 1997, 76, 121–123.
- Hammond, S.R. The Age-Range of Risk of Developing Multiple Sclerosis: Evidence from a Migrant Population in Australia. Brain 2000, 123, 968–974.
- Bach, J.-F. Revisiting the Hygiene Hypothesis in the Context of Autoimmunity. Front. Immunol. 2021, 11, 615192.
- Krassas, G.E.; Tziomalos, K.; Pontikides, N.; Lewy, H.; Laron, Z. Seasonality of Month of Birth of Patients with Graves’ and Hashimoto’s Diseases Differ from That in the General Population. Eur. J. Endocrinol. 2007, 156, 631–636.
- Attard, C.C.; Sze, W.C.C.; Vella, S. Predictors of Autoimmune Thyroid Disease. Bayl. Univ. Med. Cent. Proc. 2022, 35, 608–614.
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Chen, Y.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2011. Nucleic Acids Res. 2011, 39, D800–D806.
- Borchers, C.H.; Kast, J.; Foster, L.J.; Siu, K.W.M.; Overall, C.M.; Binkowski, T.A.; Hildebrand, W.H.; Scherer, A.; Mansoor, M.; Keown, P.A. The Human Proteome Organization Chromosome 6 Consortium: Integrating Chromosome-Centric and Biology/Disease Driven Strategies. J. Proteom. 2014, 100, 60–67.
- Kulski, J.K.; Suzuki, S.; Shiina, T. Human Leukocyte Antigen Super-Locus: Nexus of Genomic Supergenes, SNPs, Indels, Transcripts, and Haplotypes. Hum. Genome Var. 2022, 9, 49.
- Zaletel, K.; Gaberšček, S. Hashimoto’s Thyroiditis: From Genes to the Disease. Curr. Genom. 2011, 12, 576–588.
- Douillard, V.; Castelli, E.C.; Mack, S.J.; Hollenbach, J.A.; Gourraud, P.-A.; Vince, N.; Limou, S. Approaching Genetics Through the MHC Lens: Tools and Methods for HLA Research. Front. Genet. 2021, 12, 774916.
- De Santis, D.; Truong, L.; Martinez, P.; D’Orsogna, L. Rapid High-resolution HLA Genotyping by MinION Oxford Nanopore Sequencing for Deceased Donor Organ Allocation. HLA 2020, 96, 141–162.
- Zawadzka-Starczewska, K.; Tymoniuk, B.; Stasiak, B.; Lewiński, A.; Stasiak, M. Actual Associations between HLA Haplotype and Graves’ Disease Development. J. Clin. Med. 2022, 11, 2492.
- Stasiak, M.; Zawadzka-Starczewska, K.; Tymoniuk, B.; Stasiak, B.; Lewiński, A. Significance of HLA in the Development of Graves’ Orbitopathy. Genes Immun. 2023, 24, 32–38.
- Liao, W.-L.; Liu, T.-Y.; Cheng, C.-F.; Chou, Y.-P.; Wang, T.-Y.; Chang, Y.-W.; Chen, S.-Y.; Tsai, F.-J. Analysis of HLA Variants and Graves’ Disease and Its Comorbidities Using a High Resolution Imputation System to Examine Electronic Medical Health Records. Front. Endocrinol. 2022, 13, 842673.
- Mori, K.; Yoshida, K. Viral Infection in Induction of Hashimotoʼs Thyroiditis: A Key Player or Just a Bystander? Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 418–424.
- Weider, T.; Genoni, A.; Broccolo, F.; Paulsen, T.H.; Dahl-Jørgensen, K.; Toniolo, A.; Hammerstad, S.S. High Prevalence of Common Human Viruses in Thyroid Tissue. Front. Endocrinol. 2022, 13, 938633.
- Wang, J.; Zhang, W.; Liu, H.; Wang, D.; Wang, W.; Li, Y.; Wang, Z.; Wang, L.; Zhang, W.; Huang, G. Parvovirus B19 Infection Associated with Hashimoto’s Thyroiditis in Adults. J. Infect. 2010, 60, 360–370.
- Heidari, Z.; Jami, M. Parvovirus B19 Infection Is Associated with Autoimmune Thyroid Disease in Adults. Int. J. Endocrinol. Metab. 2021, 19, e115592.
- Pastore, F. Hepatitis C Virus Infection and Thyroid Autoimmune Disorders: A Model of Interactions between the Host and the Environment. WJH 2016, 8, 83.
- Caselli, E.; Zatelli, M.C.; Rizzo, R.; Benedetti, S.; Martorelli, D.; Trasforini, G.; Cassai, E.; Degli Uberti, E.C.; Di Luca, D.; Dolcetti, R. Virologic and Immunologic Evidence Supporting an Association between HHV-6 and Hashimoto’s Thyroiditis. PLoS Pathog. 2012, 8, e1002951.
- Seyyedi, N.; Dehbidi, G.R.; Karimi, M.; Asgari, A.; Esmaeili, B.; Zare, F.; Farhadi, A.; Dabbaghmanesh, M.H.; Saki, F.; Behzad-Behbahani, A. Human Herpesvirus 6A Active Infection in Patients with Autoimmune Hashimoto’s Thyroiditis. Braz. J. Infect. Dis. 2019, 23, 435–440.
- Trovato, M.; Sciacchitano, S.; Facciolà, A.; Valenti, A.; Visalli, G.; Di Pietro, A. Interleukin-6 Signalling as a Valuable Cornerstone for Molecular Medicine (Review). Int. J. Mol. Med. 2021, 47, 107.
- Trovato, M.; Ruggeri, R.M.; Sciacchitano, S.; Vicchio, T.M.; Picerno, I.; Pellicanò, G.; Valenti, A.; Visalli, G. Serum Interleukin-6 Levels Are Increased in HIV-Infected Patients That Develop Autoimmune Disease during Long-Term Follow-Up. Immunobiology 2018, 223, 264–268.
- Lin, C.-Y.; Chung, Y.-H.; Shi, Y.-F.; Tzang, B.-S.; Hsu, T.-C. The VP1 Unique Region of Human Parvovirus B19 and Human Bocavirus Induce Lung Injury in Naïve Balb/c Mice. PLoS ONE 2018, 13, e0202667.
- Canuti, M.; Eis-Huebinger, A.M.; Deijs, M.; De Vries, M.; Drexler, J.F.; Oppong, S.K.; Müller, M.A.; Klose, S.M.; Wellinghausen, N.; Cottontail, V.M.; et al. Two Novel Parvoviruses in Frugivorous New and Old World Bats. PLoS ONE 2011, 6, e29140.
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368.
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146.
- Lozano-Fernandez, J.; Carton, R.; Tanner, A.R.; Puttick, M.N.; Blaxter, M.; Vinther, J.; Olesen, J.; Giribet, G.; Edgecombe, G.D.; Pisani, D. A Molecular Palaeobiological Exploration of Arthropod Terrestrialization. Phil. Trans. R. Soc. B 2016, 371, 20150133.
- Lozano-Fernandez, J.; Tanner, A.R.; Puttick, M.N.; Vinther, J.; Edgecombe, G.D.; Pisani, D. A Cambrian–Ordovician Terrestrialization of Arachnids. Front. Genet. 2020, 11, 182.
- Pénzes, J.J.; De Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects Both Vertebrate and Invertebrate Hosts. Viruses 2019, 11, 525.
- Cossart, Y.E.; Cant, B.; Field, A.M.; Widdows, D. Parvovirus-like particles in human sera. Lancet 1975, 305, 72–73.
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a Human Parvovirus by Molecular Screening of Respiratory Tract Samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896.
- Lehmann, H.W.; Von Landenberg, P.; Modrow, S. Parvovirus B19 Infection and Autoimmune Disease. Autoimmun. Rev. 2003, 2, 218–223.
- Adamson, L.A.; Fowler, L.J.; Clare-Salzler, M.J.; Hobbs, J.A. Parvovirus B19 Infection in Hashimoto’s Thyroiditis, Papillary Thyroid Carcinoma, and Anaplastic Thyroid Carcinoma. Thyroid 2011, 21, 411–417.
- Wang, J.H.; Zhang, W.P.; Liu, H.X.; Wang, D.; Li, Y.F.; Wang, W.Q.; Wang, L.; He, F.R.; Wang, Z.; Yan, Q.G.; et al. Detection of Human Parvovirus B19 in Papillary Thyroid Carcinoma. Br. J. Cancer 2008, 98, 611–618.
- Gravelsina, S.; Nora-Krukle, Z.; Svirskis, S.; Cunskis, E.; Murovska, M. Presence of B19V in Patients with Thyroid Gland Disorders. Medicina 2019, 55, 774.
- Wang, X.; Xu, P.; Cheng, F.; Li, Y.; Wang, Z.; Hao, S.; Wang, J.; Ning, K.; Ganaie, S.S.; Engelhardt, J.F.; et al. Cellular Cleavage and Polyadenylation Specificity Factor 6 (CPSF6) Mediates Nuclear Import of Human Bocavirus 1 NP1 Protein and Modulates Viral Capsid Protein Expression. J. Virol. 2020, 94, e01444-19.
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A Critical Role for Alternative Polyadenylation Factor CPSF6 in Targeting HIV-1 Integration to Transcriptionally Active Chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063.
- Zheng, Y.; Schubert, H.L.; Singh, P.K.; Martins, L.J.; Engelman, A.N.; D’Orso, I.; Hill, C.P.; Planelles, V. Cleavage and Polyadenylation Specificity Factor 6 Is Required for Efficient HIV-1 Latency Reversal. mBio 2021, 12, e01098-21.
- Mattola, S.; Hakanen, S.; Salminen, S.; Aho, V.; Mäntylä, E.; Ihalainen, T.O.; Kann, M.; Vihinen-Ranta, M. Concepts to Reveal Parvovirus–Nucleus Interactions. Viruses 2021, 13, 1306.