Colorectal cancer (CRC) remains a major life-threatening malignancy. Apoptosis and autophagy are two processes that share common signaling pathways, are linked by functional relationships and have similar protein components. During the development of cancer, the two processes can trigger simultaneously in the same cell, causing, in some cases, an inhibition of autophagy by apoptosis or apoptosis by autophagy. Malignant cells that have accumulated genetic alterations can take advantage of any alterations in the apoptotic process and as a result, progress easily in the cancerous transformation. Autophagy often plays a suppressive role during the initial stages of carcinogenicity, while in the later stages of cancer development it can play a promoting role. It is extremely important to determine the regulation of this duality of autophagy in the development of CRC and to identify the molecules involved, as well as the signals and the mechanisms behind it. All the reported experimental results indicate that, while the antagonistic effects of autophagy and apoptosis occur in an adverse environment characterized by deprivation of oxygen and nutrients, leading to the formation and development of CRC, the effects of promotion and collaboration usually involve an auxiliary role of autophagy compared to apoptosis.
- human colorectal cancer
- apoptosis
- autophagy
1. Introduction
2. Apoptosis and Autophagy in Adenoma–Carcinoma Sequence
The crypts of the human colorectal mucosa exhibit a polarized structural organization. The reserve pool of the stem cells of the regenerative epithelium is very small and located at the base of the crypt. Stem cells move from the bottom of the crypt up, and on the apical margin of the mucosa about 10 million cells per day die from apoptosis, after which they fall into the lumen [7]. For this reason, adequate cell death is required to maintain the homeostasis of normal colorectal mucosa. However, defective signaling or unbalanced regulation of apoptosis is probably one of the causes of the onset and progression of an adenoma to the carcinoma sequence that results in CRC. It should be noted that the proteins most involved in apoptosis, such as Bcl-2 antagonist killer 1 (Bak) or Bcl-2, are not equally expressed in all anatomical regions of the colorectal mucosa, confirming the fact that there is a diversified regulation of death in the bowel [8][9]. Moreover, not only apoptosis, as a classical form of programmed cell death, has been shown to be operative and consistent in the colorectal glands, but also autophagy, a controlled process of cell self-digestion of great importance in situations of cellular stress, lack of nutrients, or energy deprivation (Figure 1) [10].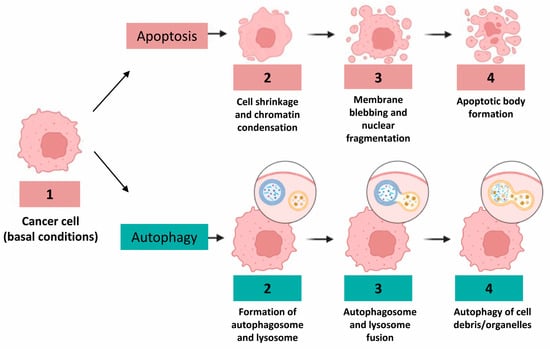
3. Regulatory Role of p53 in Apoptotic and Autophagic Processes
4. SQSTM1/p62 Is Related to Autophagy and Apoptosis in CRC Cells
SQSTM1/p62 is an autophagy receptor involved in optimizing the autophagosome formation process. After the fusion of autophagosomes with lysosomes, the autophagosome content, as well as SQSTM1/p62, is degraded [26]. SQSTM1/p62 plays specific and indispensable roles in selective autophagy and SQSTM1/p62 protein levels have been used as an indicator of autophagic flux [27]. The pivotal roles of SQSTM1/p62 in ingested protein catabolism through autophagy and lysosomal targeting have been well elucidated in the literature [26][27][28]. Although the role of SQSTM1/p62 in solid tumors is still debated, evidence has shown that SQSTM1/p62 is upregulated in different cancers and promotes tumor growth. Moreover, it has been assessed that SQSTM1/p62, as a tumor oncogene, is frequently abnormally upregulated and engaged in the acquired malignancy of gastrointestinal tumors, such as CRC [29]. Recently, Mukherjee et al. stated that, in HCT116 colorectal cells, SQSTM1/p62 is a mutp53 interactor, which associates selectively with the DNA contact mutant p53R273H but not with the structural mutant p53R175H. They further assess that the interaction with SQSTM1/p62 is needed for the ability of p53R273H to trigger cancer cell migration and invasion. The scholars assumed that this acquired ability is due to the involvement of the mutp53-p62 axis in directing the ubiquitin-dependent proteasome degradation of connexin 43 and cell junction proteins [30]. Nevertheless, the autophagic process can be repressed by SQSTM1/p62 inhibition, leading to the arrest of cancer cell growth and the inhibition of cancer development [31]. On the other hand, literature has reported that SQSTM1/p62 expression in CRC cells was suppressed by the β-catenin/transcription factor (TCF)4 complex, which blocked phagocytic ingestion, but under stress conditions induced by nutrient depletion, SQSTM1/p62 was markedly raised, as β-catenin was immediately degraded by LC3 binding to the LC3-interacting region (LIR) of β-catenin [31].5. NF-κB as a Potential Matchmaker between Autophagy and Apoptosis
Studies on CRC have reported that the NF-kB signaling pathway promotes tumor initiation and contributes to cancer cell metastasis formation and epithelial to mesenchymal transition [32][33][34]. In the literature, there is a plethora of papers that confirm the role of NF-kB in regulating the apoptotic process. In fact, the molecular mechanisms by which NF-kB exerts its regulatory action are well known, for instance: NF-κB signaling prevents apoptosis by up-regulating anti-apoptotic genes expression such as B-cell lymphoma-extra-large (Bcl-xL), the Bcl-2-related gene (A1/BFL1), cellular inhibitors of apoptosis proteins (cIAPs) and caspase-8/FAS-associated death domain-like IL-1beta-converting enzyme inhibitory protein (c-FLIP) [33]. Over the past few years, some very interesting studies have highlighted a correlation between NF-kB, autophagy and apoptotic processes. A study showed that metformin had an antiproliferative effect related to changes in the expression of nuclear factor E2-related factor (NRF-2)/NF-κB pathways on human colon cancer cells (HT-29) in a dose- and time-dependent manner, and exerted growth inhibitory effects by increasing both apoptosis and autophagy [35].Recently, the autophagy-regulated cytotoxic effect of green synthesized silver nanoparticles (Brassica Ag-NPs) against human epithelial colorectal adenocarcinoma cells was demonstrated. The scholars found that Brassica Ag-NPs induced NF-κB mediated autophagy in Caco-2 cells. The decreased expression of NFκB was associated with an increased expression of inhibitor of NF-κB (IκB)-kinase, which is involved in autophagic process initiation. Moreover, the activity of p53 and light chain 3 (LC3) II greatly accelerated autophagosome formation, and the inhibition of Akt and mammalian target of rapamycin (mTOR) was evident. In conclusion, the scholars stated that, in the event of excessive activation of the autophagic process, after the complete depletion of nutrients, this process stalled, leading to the accumulation of aberrant metabolic intermediates that can trigger apoptotic cell death, which subsequently resulted in necrosis [36].
6. Oxidative Stress and Modulation of Autophagy and Apoptosis
Oxidative stress has been implicated in the pathophysiology of cancer: high levels of intra-cytoplasmatic reactive oxygen species (ROS), generated by accelerate aerobic glycolysis followed by “selfish” metabolic reprogramming (the Warburg effect), increase oncogene activity, the activation of growth factor dependent pathways or the presence of an increase in the pool of oxidizing enzymes, leads to genetic instability [37][38]. Aerobic glycolysis or the Warburg effect is a hallmark of metabolic phenotypes of cancer; in fact, cancer cells, including CRC cells, present altered glucose metabolism and are characterized by an enhanced uptake of glucose and an increased conversion of glucose to lactate [39]. In the past few years, many studies have focused on the importance of the role played by ROS in the crosstalk between autophagy and apoptosis of colon cancer cells. ROS has a negative feedback action on the autophagic process: ROS can promote autophagy, and at the same time, autophagy reduces ROS production by removing damaged mitochondria, endoplasmic reticulum and other materials that contribute to ROS generation [40][41]. On the other hand, ROS can trigger DNA damage, initiating the apoptotic process pathway in colon cancer cells [42][43]. The most common method by which ROS delete transformed cells is the activation of programmed cell death, which is completed by an extrinsic or an intrinsic pathway; both pathways culminate in caspase-induced final cell demise with the formation of apoptotic bodies that are removed by adjacent phagocytes (Figure 24) [44].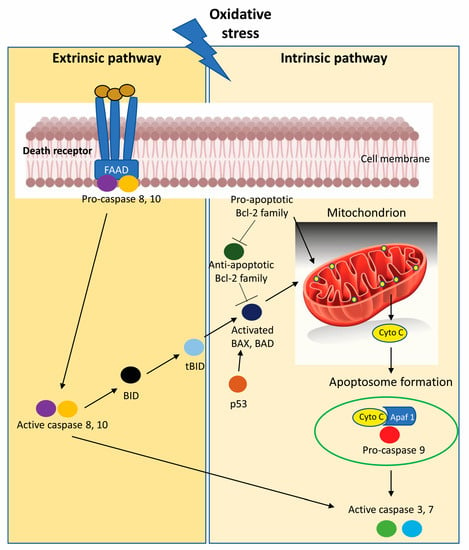
7. Role of Non-Coding RNA in Enhancing or Inhibiting Autophagy and Apoptosis
8. Pathways Implicated in Regulation of Apoptosis and Autophagy and Possible Therapeutic Targets
The most important pathway involved in the induction and regulation of apoptosis and autophagy in CRC is the Wnt pathway and its component components, such as β-catenin, disheveled (Dvl), adenomatous polyposis coli (APC) and axin, that are activated along with the Wnt signaling cascade during cancer development [56]. Wnt signal transduction is typically divided into classical and non-classical pathways. The classical pathway is involved in cell survival, proliferation, apoptosis and autophagy, while the non-classical pathway regulates cell polarity and migration. The Wnt/β-catenin signaling and autophagy pathways play important roles in tissue homeostasis and tumorigenesis. A variety of studies, reporting experimental design of the genomic manipulation of β-catenin expression levels in vitro and in vivo, show that β-catenin inhibits autophagosome formation and directly blocks SQSTM1/p62 via T cell-specific DNA-binding protein (TCF4) [57]. During starvation, β-catenin is selectively degraded through the formation of β-catenin-LC3 complexes, which avoid β-catenin/T cell-specific DNA-binding protein (TCF) mediated transcription and proliferation in order to better adapt to metabolic stress conditions. The formation of the β-catenin-LC3 complex is mediated by the W/YXXI/L motif and the LC3 interaction region (LIR) in β-catenin. Therefore, as previously described, Wnt/β-catenin inhibits autophagy and SQSTM1/p62 expression, while β-catenin itself is targeted for autophagic degradation [58]. Autophagy negatively controls Wnt signaling by promoting Dvl degradation. Von Hippel–Lindau protein-mediated ubiquitination is necessary for the binding of Dvl2 to SQSTM1/p62, leading to the formation of Dvl2 aggregates under conditions of nutrient deprivation and LC3-mediated autophagosome recruitment. Finally, the ubiquitylated Dvl2 aggregates are degraded through the autophagy-lysosome pathway [59]. Interestingly, an inverse correlation between Dvl expression and autophagy was found in late stages of CRC development, supporting the hypothesis that autophagy may contribute to the aberrant promotion of Wnt signaling during tumorigenesis [56].9. Conclusions
While the antagonistic effects of autophagy and apoptosis occur in an adverse environment characterized by deprivation of oxygen and nutrients leading to the formation and development of CRC, the effects of promotion and collaboration usually involve an auxiliary role of autophagy compared to apoptosis in colon cancer cells. In an overwhelming majority of cases, studies examining the mechanistic role of autophagy and apoptosis on CRC tumorigenesis used xenograft mice models and CRC cell lines. Moreover, many of these studies have targeted autophagy and apoptosis transiently through genetic manipulations or the use of drugs, which can influence the study in various ways. From our point of view, it would be necessary for preclinical research to focus on the improvement of gene-editing techniques, which are necessary to delineate the mechanistic role of autophagy and apoptosis in CRC. The results obtained at this stage would be of great use in clinical trials in patients to test the response to immune-checkpoint inhibitors. In addition, it is necessary to focus attention on ongoing clinical trials that are using apoptosis or autophagy inhibitors in combination with chemotherapeutics or drugs. These studies aim to capture the positive results of using autophagy as a process of inducing programmed cell death in cancer cells. Such autophagic manipulation can lead to promising effects in the treatment of CRC if it will attempt to solve the autophagy paradox that migrates from an anti-cancer to a pro-cancer mechanism, so treatments need to be highly specific for the given setting.References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249.
- Leslie, A.; Carey, F.A.; Pratt, N.R.; Steele, R.J.C. The Colorectal Adenoma–Carcinoma Sequence. Br. J. Surg. 2002, 89, 845–860.
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The Landscape of Somatic Mutation in Normal Colorectal Epithelial Cells. Nature 2019, 574, 532–537.
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and Apoptotic Body: Disease Message and Therapeutic Target Potentials. Biosci. Rep. 2019, 39, BSR20180992.
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054.
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592.
- McCarthy, N.; Manieri, E.; Storm, E.E.; Saadatpour, A.; Luoma, A.M.; Kapoor, V.N.; Madha, S.; Gaynor, L.T.; Cox, C.; Keerthivasan, S.; et al. Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell Stem Cell 2020, 26, 391–402.e5.
- Duckworth, C.A.; Pritchard, D.M. Suppression of Apoptosis, Crypt Hyperplasia, and Altered Differentiation in the Colonic Epithelia of Bak-Null Mice. Gastroenterology 2009, 136, 943–952.
- Qiao, L.; Wong, B.C.Y. Targeting Apoptosis as an Approach for Gastrointestinal Cancer Therapy. Drug Resist. Updates 2009, 12, 55–64.
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The Role of Autophagy in Colitis-Associated Colorectal Cancer. Signal Transduct. Target. Ther. 2018, 3, 31.
- Finkbeiner, S. The Autophagy Lysosomal Pathway and Neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a033993.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339.
- Hasbullah, H.H.; Musa, M. Gene Therapy Targeting P53 and KRAS for Colorectal Cancer Treatment: A Myth or the Way Forward? Int. J. Mol. Sci. 2021, 22, 11941.
- Nakayama, M.; Oshima, M. Mutant P53 in Colon Cancer. J. Mol. Cell Biol. 2019, 11, 267–276.
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2020, 22, 130.
- Nishiumi, S.; Fujishima, Y.; Inoue, J.; Masuda, A.; Azuma, T.; Yoshida, M. Autophagy in the Intestinal Epithelium Is Not Involved in the Pathogenesis of Intestinal Tumors. Biochem. Biophys. Res. Commun. 2012, 421, 768–772.
- Wang, L.; Wang, Y.; Lu, Y.; Zhang, Q.; Qu, X. Heterozygous Deletion of ATG5 in Apc Min/+ Mice Promotes Intestinal Adenoma Growth and Enhances the Antitumor Efficacy of Interferon-Gamma. Cancer Biol. Ther. 2015, 16, 383–391.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Hu, B.; Yin, Y.; Li, S.; Guo, X. Insights on Ferroptosis and Colorectal Cancer: Progress and Updates. Molecules 2022, 28, 243.
- Dong, Y.; Wu, Y.; Zhao, G.-L.; Ye, Z.-Y.; Xing, C.-G.; Yang, X.-D. Inhibition of Autophagy by 3-MA Promotes Hypoxia-Induced Apoptosis in Human Colorectal Cancer Cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1047–1054.
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fåhraeus, R. The P53 MRNA: An Integral Part of the Cellular Stress Response. Nucleic Acids Res. 2019, 47, 3257–3271.
- Yamada, K.; Yoshida, K. Mechanical Insights into the Regulation of Programmed Cell Death by P53 via Mitochondria. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 839–848.
- Shajani-Yi, Z.; de Abreu, F.B.; Peterson, J.D.; Tsongalis, G.J. Frequency of Somatic TP53 Mutations in Combination with Known Pathogenic Mutations in Colon Adenocarcinoma, Non–Small Cell Lung Carcinoma, and Gliomas as Identified by Next-Generation Sequencing. Neoplasia 2018, 20, 256–262.
- Sakanashi, F.; Shintani, M.; Tsuneyoshi, M.; Ohsaki, H.; Kamoshida, S. Apoptosis, Necroptosis and Autophagy in Colorectal Cancer: Associations with Tumor Aggressiveness and P53 Status. Pathol. Res. Pract. 2019, 215, 152425.
- Gurkan, A.C.; Arisan, E.D.; Yerlikaya, P.O.; Ilhan, H.; Unsal, N.P. Inhibition of Autophagy Enhances DENSpm-Induced Apoptosis in Human Colon Cancer Cells in a P53 Independent Manner. Cell. Oncol. 2018, 41, 297–317.
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of Selective Autophagy: The P62/SQSTM1 Paradigm. Essays Biochem. 2017, 61, 609–624.
- Salazar, G.; Cullen, A.; Huang, J.; Zhao, Y.; Serino, A.; Hilenski, L.; Patrushev, N.; Forouzandeh, F.; Hwang, H.S. SQSTM1/P62 and PPARGC1A/PGC-1alpha at the Interface of Autophagy and Vascular Senescence. Autophagy 2020, 16, 1092–1110.
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224.
- Kosumi, K.; Masugi, Y.; Yang, J.; Qian, Z.R.; Kim, S.A.; Li, W.; Shi, Y.; da Silva, A.; Hamada, T.; Liu, L.; et al. Tumor SQSTM1 (P62) Expression and T Cells in Colorectal Cancer. OncoImmunology 2017, 6, e1284720.
- Mukherjee, S.; Maddalena, M.; Lü, Y.; Martinez, S.; Nataraj, N.B.; Noronha, A.; Sinha, S.; Teng, K.; Cohen-Kaplan, V.; Ziv, T.; et al. Cross-Talk between Mutant P53 and P62/SQSTM1 Augments Cancer Cell Migration by Promoting the Degradation of Cell Adhesion Proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2119644119.
- Tang, J.; Li, Y.; Xia, S.; Li, J.; Yang, Q.; Ding, K.; Zhang, H. Sequestosome 1/P62: A Multitasker in the Regulation of Malignant Tumor Aggression (Review). Int. J. Oncol. 2021, 59, 77.
- Plewka, D.; Plewka, A.; Miskiewicz, A.; Morek, M.; Bogunia, E. Nuclear Factor-Kappa B as Potential Therapeutic Target in Human Colon Cancer. J. Can. Res. Ther. 2018, 14, 516.
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-ΚB Signaling Pathway in the Pathogenesis of Colorectal Cancer. Gene 2020, 726, 144132.
- Xin, J. Critical Signaling Pathways Governing Colitis-Associated Colorectal Cancer: Signaling, Therapeutic Implications, and Challenges. Dig. Liver Dis. 2023, 55, 169–177.
- Sena, P.; Mancini, S.; Benincasa, M.; Mariani, F.; Palumbo, C.; Roncucci, L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. Int. J. Mol. Sci. 2018, 19, 1478.
- Akter, M.; Atique Ullah, A.K.M.; Banik, S.; Sikder, M.T.; Hosokawa, T.; Saito, T.; Kurasaki, M. Green Synthesized Silver Nanoparticles-Mediated Cytotoxic Effect in Colorectal Cancer Cells: NF-ΚB Signal Induced Apoptosis Through Autophagy. Biol. Trace Elem. Res. 2021, 199, 3272–3286.
- Vaupel, P.; Multhoff, G. Revisiting the Warburg Effect: Historical Dogma versus Current Understanding. J. Physiol. 2021, 599, 1745–1757.
- Keller, F.; Bruch, R.; Schneider, R.; Meier-Hubberten, J.; Hafner, M.; Rudolf, R. A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro. Cells 2020, 9, 1900.
- Kasprzak, A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434.
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, Mitochondria and Oxidative Stress: Cross-Talk and Redox Signalling. Biochem. J. 2012, 441, 523–540.
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative Stress, Redox Signaling, and Autophagy: Cell Death Versus Survival. Antioxid. Redox Signal. 2014, 21, 66–85.
- Yu, G.; Luo, H.; Zhang, N.; Wang, Y.; Li, Y.; Huang, H.; Liu, Y.; Hu, Y.; Liu, H.; Zhang, J.; et al. Loss of P53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation. Int. J. Mol. Sci. 2019, 20, 6268.
- da Silva, E.L.; Mesquita, F.P.; Ramos, I.N.d.F.; Gomes, C.B.d.S.M.R.; Moreira, C.d.S.; Ferreira, V.F.; da Rocha, D.R.; Bahia, M.d.O.; Moreira-Nunes, C.A.; de Souza, C.R.T.; et al. Antitumoral Effect of Novel Synthetic 8-Hydroxy-2-((4-Nitrophenyl)Thio)Naphthalene-1,4-Dione (CNN16) via ROS-Mediated DNA Damage, Apoptosis and Anti-Migratory Effect in Colon Cancer Cell Line. Toxicol. Appl. Pharmacol. 2022, 456, 116256.
- Gao, L.; Loveless, J.; Shay, C.; Teng, Y. Targeting ROS-Mediated Crosstalk Between Autophagy and Apoptosis in Cancer. In Reviews on New Drug Targets in Age-Related Disorders; Guest, P.C., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1260, pp. 1–12. ISBN 978-3-030-42666-8.
- Lee, S.-J.; Lee, D.-E.; Choi, S.-Y.; Kwon, O.-S. OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-ΚB Signaling in Colon Cancer Cells. Int. J. Mol. Sci. 2021, 22, 11073.
- Shi, W.; Men, L.; Pi, X.; Jiang, T.; Peng, D.; Huo, S.; Luo, P.; Wang, M.; Guo, J.; Jiang, Y.; et al. Shikonin Suppresses Colon Cancer Cell Growth and Exerts Synergistic Effects by Regulating ADAM17 and the IL-6/STAT3 Signaling Pathway. Int. J. Oncol. 2021, 59, 99.
- Ding, R.; Ning, X.; Ye, M.; Yin, Y. Antrodia Camphorata Extract (ACE)-Induced Apoptosis Is Associated with BMP4 Expression and P53-Dependent ROS Generation in Human Colon Cancer Cells. J. Ethnopharmacol. 2021, 268, 113570.
- Hong, H.; Cao, W.; Wang, Q.; Liu, C.; Huang, C. Synergistic Antitumor Effect of Andrographolide and Cisplatin through ROS-Mediated ER Stress and STAT3 Inhibition in Colon Cancer. Med. Oncol. 2022, 39, 101.
- Choi, Y.J.; Lee, J.; Ha, S.H.; Lee, H.K.; Lim, H.M.; Yu, S.; Lee, C.M.; Nam, M.J.; Yang, Y.; Park, K.; et al. 6, 8-Diprenylorobol Induces Apoptosis in Human Colon Cancer Cells via Activation of Intracellular Reactive Oxygen Species and P53. Environ. Toxicol. 2021, 36, 914–925.
- Zhang, N.; Hu, X.; Du, Y.; Du, J. The Role of MiRNAs in Colorectal Cancer Progression and Chemoradiotherapy. Biomed. Pharmacother. 2021, 134, 111099.
- Zhang, J.; Yang, W.; Xiao, Y.; Shan, L. MiR-125b Inhibits Cell Proliferation and Induces Apoptosis in HumanColon Cancer SW480 Cells via Targeting STAT3. Recent Pat. Anti-Cancer Drug Discov. 2022, 17, 187–194.
- Gao, J.; Fei, L.; Wu, X.; Li, H. MiR-766-3p Suppresses Malignant Behaviors and Stimulates Apoptosis of Colon Cancer Cells via Targeting TGFBI. Can. J. Gastroenterol. Hepatol. 2022, 2022, 7234704.
- Chen, S.; Wang, Y.; Xu, M.; Zhang, L.; Su, Y.; Wang, B.; Zhang, X. MiR-1184 Regulates the Proliferation and Apoptosis of Colon Cancer Cells via Targeting CSNK2A1. Mol. Cell. Probes 2020, 53, 101625.
- Liao, D.; Li, T.; Ye, C.; Zeng, L.; Li, H.; Pu, X.; Ding, C.; He, Z.; Huang, G. MiR-221 Inhibits Autophagy and Targets TP53INP1 in Colorectal Cancer Cells. Exp. Ther. Med. 2017, 15, 1712–1717.
- Zhang, R.; Xu, J.; Zhao, J.; Bai, J. Mir-30d Suppresses Cell Proliferation of Colon Cancer Cells by Inhibiting Cell Autophagy and Promoting Cell Apoptosis. Tumour Biol. 2017, 39, 101042831770398.
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt Signaling in Colorectal Cancer: Pathogenic Role and Therapeutic Target. Mol. Cancer 2022, 21, 144.
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-Catenin Signalling: Function, Biological Mechanisms, and Therapeutic Opportunities. Signal Transduct. Target. Ther. 2022, 7, 3.
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal β-Catenin Degradation Regulates Wnt-Autophagy-P62 Crosstalk. EMBO J. 2013, 32, 1903–1916.
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy Negatively Regulates Wnt Signalling by Promoting Dishevelled Degradation. Nat. Cell. Biol. 2010, 12, 781–790.