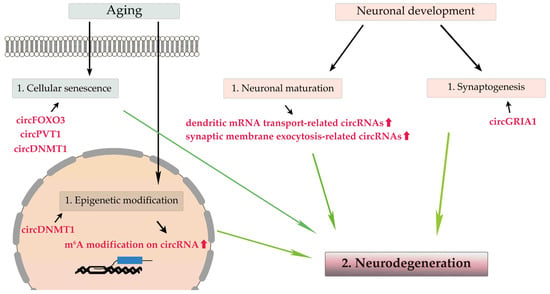
During neuronal maturation, the expression levels of most circRNAs, especially cognate mRNAs that are translated into proteins related to dendritic mRNA transport and synaptic membrane exocytosis, are upregulated without correlation to their cognate RNAs’ expression levels
. Additionally, during synaptogenesis, many circRNAs expressed in the brain are enriched in the synaptic space, and their expression levels are altered regardless of their cognate linear mRNAs’ expression levels
. Therefore, circRNAs play a pivotal role in neuronal maturation and synaptogenesis independent of their cognate mRNAs
, and perturbation of circRNA expression may be a cause of neurodegeneration.
Among the three ADAR proteins expressed in mammals, ADAR1 and ADAR2 have essential editing activities. ADAR1 is ubiquitously expressed and is primarily responsible for RNA editing in repeat elements in noncoding regions of mRNAs, whereas ADAR2, which is expressed highest in the CNS, is involved in recoding and editing in protein-coding regions
[21][47][36,70]. ADAR3, which is highly enriched in oligodendrocytes in the brain, lacks editing activity and instead acts as a negative regulator of RNA editing by sequestering the editing substrates of ADAR1 and ADAR2
[21][48][49][36,71,72]. RNA editing within protein-coding sequences alters protein structure and function, leading to alterations in multiple biological processes, including synaptic transmission and immune responses
[22][50][51][37,73,74]. RNA editing is increased during development in the mammalian brain
[52][53][75,76] and, conversely, decreased in age-related diseases
[54][55][77,78]. These findings suggest a modulatory role for RNA editing in human aging.
3. ALS-Related Changes of circRNAs and Dysregulation of RNA Editing
3.1. ALS-Related circRNA
CircRNAs are associated with brain aging and neurodegeneration, and the presence of disease-specific circRNAs has been reported in the brains of patients with Alzheimer’s or Parkinson’s disease
[56][84] as well as in the spinal cord and muscles of patients with ALS
[57][58][85,86].
Several ALS-linked genes encode RBPs, and the aberrant protein aggregation of RBPs is a pathological characteristic of motor neurons in ALS. Therefore, the resulting disruption of RNA metabolism plays a key role in ALS pathogenesis
[6]. FUS plays a critical role in splicing regulation, and subcellular mislocalization of FUS leads to cell death-causing aberrant RNA metabolism in ALS motor neurons
[59][87]. The biogenesis of circRNAs is affected by the interaction of FUS with intron-flanking back-splicing junctions without significant effects on the expression of cognate linear RNAs
[60][88]. Additionally, the expression levels of several circRNAs are altered in induced pluripotent stem cell (iPSC)-derived motor neurons of patients with ALS carrying the
FUSP525L mutation compared with those carrying
FUSWT [61][89], although the pathogenic significance and effects on motor neuron biology of the circRNA expression changes in ALS patients carrying the
FUS mutation remain unknown. The intronic hexanucleotide (GGGGCC) repeat expansion (HRE) of
C9orf72 is the most common genetic cause of ALS in Europe and America
[62][63][90,91] in which pathogenic roles of non-AUG translation-mediated production of toxic dipeptide repeat (DPR) proteins and sequestration of RBP in nuclear RNA granules have been hypothesized
[64][92].
Evidence suggests a role for circRNAs in the epigenetic modification of nucleic acids in ALS
[65][94]. Methyl-CpG binding domain protein 2 (MDB2) binds to a fraction of hypomethylated genes and plays a pivotal role in methylation-related transcription regulation
[66][95]. Knockdown of circKCNN2 (has_circ_0127664), a circular form of potassium calcium-activated channel subfamily N member 2 (
KCNN2), leads to the downregulation of MDB2
[67][96]. Expression levels of circKCNN2 are considerably reduced in the cortical neurons of patients with frontotemporal dementia, exhibiting mislocalization of the transactive response DNA-binding protein of 43 kDa (TDP-43) from the nucleus to the cytoplasm (TDP-43 pathology) as compared with those in control subjects
[68][97].
A pathogenic role for excitotoxicity resulting from excessive Ca
2+ influx into motor neurons by GluA2-lacking Ca
2+-permeable AMPA receptor ion channels has been proposed in ALS
[69][70][106,107]. AMPA receptors are homo- or hetero-tetramers of GluA1–GluA4 subunits. Their tightly regulated biogenesis, membrane trafficking, and degradation result in well-regulated physiological CNS activity
[71][108]. The upregulation of GluA1 mRNA, which reduces the proportion of the GluA2 subunit among the four subunits, is associated with excitotoxicity in the spinal cord and iPSC-derived motor neurons of patients with
C9orf72 ALS
[72][109] and
FUS knockdown mice
[73][74][75][76][110,111,112,113]. CircGRIA1 negatively regulates the expression levels of GluA1 mRNA and protein expression by competitively binding to the promotor region of
GRIA1 [77][67].
The dysfunction of the ER and mitochondria due to the alteration of ER–mitochondrial signaling is another hypothesis for ALS pathogenesis
[78][114]. The ER physically contacts the mitochondria through specialized lesions called mitochondria-associated membranes (MAMs), and studies have reported an association between MAM disruption and the pathogenesis of various neurodegenerative diseases, including ALS
[79][80][81][115,116,117]. Mutations in sigma nonopioid intracellular receptor 1 (
SIGMAR1), which encodes the sigma-1 receptor (Sig1R), cause juvenile ALS (ALS16), and mutant Sig1R loses its MAM-specific chaperone protein function
[82][118]. Overexpression of
Sig1RE102Q mutant proteins induces neuronal cell death
[83][119], and loss of wild-type Sig1R proteins induces the collapse of MAMs in the motor neurons of
Sig1R−/− mice
[79][115]. The significance of changes in the expression levels of SigR1 in ALS has been inconsistently reported; expression levels of mutant Sig1R proteins (c672*51G > T) are either elevated in leukocytes and the frontal cortex
[84][120] or not different in primary lymphoblastoid cells derived from patients with ALS carrying mutant
Sig1RE102Q [85][121].
3.2. Dysregulation of RNA Editing in ALS Motor Neurons
3.2. Dysregulation of RNA Editing in ALS Motor Neurons (Table 1)
Evidence that elevated glutamate levels in the postmortem tissue and CSF of patients with ALS
[86][87][88][89][124,125,126,127], the loss of high-affinity glutamate uptake
[90][128], and riluzole, an inhibitor of glutamate release, improve one-year survival rates, especially in the late stages of ALS
[91][92][93][94][8,129,130,131] has implicated excitotoxicity as a cause of ALS pathogenesis. Among the subtypes of glutamate receptors, Ca
2+-permeable AMPA receptors specifically mediate the slow death of motor neurons, and the increase in their Ca
2+ permeability results from the incorporation of the Q/R site-unedited GluA2 subunit into their assembly.In the spinal motor neurons of patients with sporadic ALS, Q/R site-unedited GluA2 is expressed because of the downregulation of ADAR2
[95][96][135,136]. Motor neuron-specific conditional ADAR2 knockout mice (ADAR2
flox/flox/VAChT. Cre; AR2 mice) exhibit progressive motor dysfunction with degeneration of motor neurons, resulting from excessive Ca
2+ influx into motor neurons through Ca
2+-permeable AMPA receptors that have Q/R site-unedited GluA2 subunits
[97][98][99][137,138,139]. The death cascade initiated by ADAR2 downregulation is specific to the motor neurons of patients with ALS and is not observed in other neurons of patients with ALS or in the motor neurons of normal control subjects or patients with other neurological diseases
[95][96][100][135,136,140].
3.3. Aging, circRNAs, and RNA Editing in ALS
Aging is a major risk factor for neurodegenerative diseases, and dysregulation of circRNAs and RNA editing with aging are associated with neurodegenerative diseases, including ALS. Although evidence has demonstrated age-associated changes in RNA editing activity and circRNA processing as described above, only some evidence has demonstrated RNA editing of the exonic region of circRNAs or the role of RNA editing in the biogenesis of circRNAs.
A-to-I RNA editing influences the biogenesis of circRNAs, and the complementary sequence across flanking introns, which contains many Alu repeats, facilitates the formation of circRNAs. Editing sites in Alu repeats are the main targets of ADARs
[101][102][103][147,148,149]. The expression levels of circRNAs correlate negatively with those of ADAR1 during neuronal differentiation without modulation of cognate RNAs
[15][25][104][105][106][23,40,150,151,152].
Although a large fraction of brain circRNA is derived from the exonic coding region
[15][23], whether the A-to-I sites in circRNAs are edited by ADARs, similarly to those in their cognate RNAs, is unclear. It confirmed that the editing efficiency at the Q/R site of circGRIA2 (has_circ_0125620), a circular form of
GRIA2, changed in parallel with that of the cognate GluA2 mRNA in cultured cells
[107][153]. Therefore, RNA editing of the exonic region of circRNAs may be reduced as RNA editing of their cognate mRNA is dysregulated in motor neurons in sporadic ALS.
4. CircRNAs and the Dysregulation of RNA Editing as Potential Biomarkers and Therapeutic Targets in ALS
4.1. Potential Biomarker Candidates for ALS
CircRNAs are most abundant in the brain and are stable after secretion into body fluids because of their distinctive structure
[16][45][108][24,31,155]. As dysregulation of RNA editing increases the formation of circRNAs and extracellular total circRNA levels
[107][109][153,154], changes in the expression levels of circRNAs could be biomarker candidates for ALS. Several studies have reported comprehensive changes in circRNA expression levels in tissues and sera derived from patients with ALS
[57][58][110][85,86,156]. A study has reported reduced expression levels of circPICALM (hsa_circ_0023919) and increased expression levels of circSETD3 (hsa_circ_0000567), circFAM120A (hsa_circ_0005218), circHERC1 (hsa_circ_0035796), circTAF15 (hsa_circ_0043138), circ TNRC6B (hsa_circ_0063411), and circSUSD1 (hsa_circ_0088036) in leukocytes from patients with ALS as compared with healthy control subjects
[110][156]. Among these, hsa_circ_0000567 and hsa_circ_0063411 contain binding sites for miR-9 and miR-641, respectively, the expression levels of which are shown to change in ALS patients
[111][112][157,158], and hsa_circ_0023919, hsa_circ_0063411, and hsa_circ_0088036 are potential diagnostic biomarkers because of their high sensitivity and specificity to ALS
[110][113][156,159].
4.2. Therapeutic Targets for ALS
A potential role for circRNAs in neurodegeneration has been proposed
[114][32], and a circRNA-based therapeutic strategy, such as the delivery or knockdown of circRNAs, has been put forward for ALS. Based on the hypothesis that accumulated TDP-43 is toxic to neurons, intron-derived circRNAs resulting from the inhibition of the intron debranching enzyme (DBR1), which catalyzes the debranching of lariat introns, were tested, and inhibition of DBR1 was found to suppress the toxicity of TDP-43 in yeast
[115][160]. Moreover, DNA methyl-transferases (DNMTs) inhibitor improves motor function and extends the lifespan of superoxide dismutase 1 (
SOD-1) mutant ALS model mice
[116][161], in which expression levels of DNMT1 are increased in the spinal cord. Recently, improved expression levels of circRNAs via the extracellular vesicle-mediated delivery of circRNAs were demonstrated
[117][118][162,163]; therefore, the delivery of circRNAs or siRNA-mediated knockdown to improve the expression levels of circRNAs has been developed as a potential future therapeutic strategy.
The dysregulation of RNA editing due to ADAR2 downregulation can also be a potential therapeutic target for ALS. As the downregulation of ADAR2 explains many aspects of disease-specific pathological changes in sporadic ALS, the restoration of ADAR2 activity and the reduction of excessive Ca
2+ influx through abnormal AMPA are promising therapeutic strategies for ALS. The delivery of ADAR2 cDNA using a neuron-specific promoter to motor neurons with adeno-associated virus serotype 9 (AAV-9) in AR2 mice, a mouse model of sporadic ALS, markedly suppressed progressive motor dysfunction without adverse effects
[119][164].