In humans, a total of 12 galectins have been identified. These galectins play important roles in controlling immune responses within the tumour microenvironment (TME) and the infiltration of immune cells, including different subsets of T cells, macrophages, and neutrophils, to fight against cancer cells. However, these infiltrating cells also have repair roles and are hijacked by cancer cells for pro-tumorigenic activities. Upon a better understanding of the immunomodulating functions of galectin-3 and -9, their inhibitors, namely, GB1211 and LYT-200, have been selected as candidates for clinical trials. The use of these galectin inhibitors as combined treatments with current immune checkpoint inhibitors (ICIs) is also undergoing clinical trial investigations. Through their network of binding partners, inhibition of galectin have broad downstream effects acting on CD8+ cytotoxic T cells, regulatory T cells (Tregs), Natural Killer (NK) cells, and macrophages as well as playing pro-inflammatory roles, inhibiting T-cell exhaustion to support the fight against cancer cells.
1. Introduction
The function of the immune system in fighting cancer cells has been of long-standing interest in the context of cancer therapies
[1], with research dating back to William B. Coley’s study in 1819
[2]. Since the first ICI, Cytotoxic-T-lympocyte-antigen-4 (CTLA-4), also known as ipilimumab (Yervoy), was tested and approved for the treatment of metastatic melanoma in 2015
[3][4][3,4], the number of checkpoint inhibitors has increased. In particular, when programmed cell death/ligand-1 (PD-1/PD-L1) immune checkpoint proteins are identified, these checkpoint inhibitors are now the standard regimen for immuno-oncology (I-O) therapy when tackling different solid tumours
[5]. The most common PD-1 and PD-L1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), cemiplimab (Libtayo), atezolizumab (Tecentriq), nivolumab (Bavencio), and durvalumab (Imfinzi)
[6]. In addition, ipilimumab (Yervoy) is the most common CTLA-4 inhibitor. However, the response rates (RRs) of this approach vary according to types and lines of treatment
[7]. It has been found to have a good-to-moderate response, with over a 50% RR against classic Hodgkin’s lymphoma, melanoma, and first-line combination-treated non-small cell lung cancer (NSCLC). However, some cancers, such as extensive-stage small cell lung cancer (SCLC), hepatocellular carcinoma (HCC), PD-L1
+ gastric (gastroesophageal junction type), and cervical cancers, have shown less than a 25% RR.
The current challenges to the efficiency of I-O therapies include the exhaustion of cytotoxic T cells
[8] and the need to increase the subpopulation of Tregs and other immune cells during immunosuppression
[9]. To combat these challenges, other immunomodulators have been identified, such as T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT). TIGIT is found in T cells, NK cells, and tumour cells. The mechanism of immune inactivation may occur through ITIM-dependent negative pathways
[10]. Numerous TIGIT antibodies, including BMS-986207, tiragolumab, and vibostolimab, have been developed and tested in clinical trials
[11]. The recent results of these trials, such as CITYSCRAPE
[12], have generated additional interest in investigating any novel immunosuppressors and their interacting protein mechanisms. The results of the subsequent phase III trials, such as the SKYSCRAPER series (NCT04619707, NCT04513925, NCT046665843, etc.), will need further analysis to enhance this approach to treatment. A list of this series of studies was summarised by Brazel et al. (2023)
[13].
Recently, galectins have been identified as immunomodulators
[14], joining TIGIT as new potential targets for immunotherapies. The potential candidate reagents and ongoing trial studies are listed in
Table 1. In particular, newly developed reagents, such as GB1211
[15], a galectin-3 small-molecule inhibitor, and LYT-200, an anti-galectin-9 humanised antibody, are currently on trial (NCT05240131/GALLANT-1 and NCT04666688, respectively). The safety of GB1211 has also been reported, with limited grade 1 and grade 2 adverse effects in healthy participants (NCT03809052)
[16]. The efficiency of these candidates in the current trial studies will further support the use of galectins in cancer treatments in combination with PD-1/PD-L1 and TIGIT. Other galectin inhibitors, such as OTX008 and ProLectin-M, have also been investigated in mouse animal model studies
[17][18][19][20][17,18,19,20].
Table 1.
Clinical trial studies (extracted from
, accessed on 11 May 2023).
2. Human Galectins
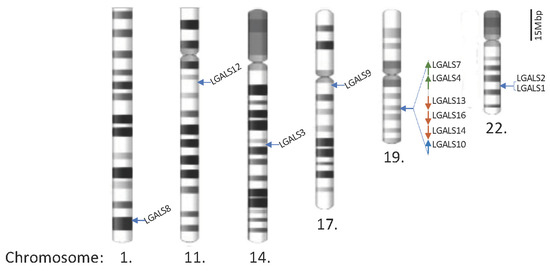
There are 12 galectins in humans, as listed in
Table 2. The genomic locations of these genes and their protein product structures are shown in
Figure 1 and
Figure 2, respectively. Galectins can be detected in the cytoplasm and exhibit a secretory form. Protein topology analysis
[24] has shown that conventional secretory proteins, which contain an N-terminal signal peptide that starts with a few positively charged amino acids, such as lysine (K) and arginine (I), followed by around 12–16 hydrophobic amino acids
[25], are driven into the endoplasmic reticulum (ER). The secretory proteins are then embedded inside lipid bilayer vesicles and transported via budding-off into the Golgi apparatus, after which the vesicles can be further fused with the cell membrane to export the protein outside the cell
[26]. However, further analysis of the coding and protein sequence of galectins has resulted in no signal peptide sequence being detected. Their secretory forms are suspected to be produced through the non-canonical secretion pathway
[27][28][27,28]. Unlike other proteins, their recognition is not based on protein peptides in the form of amino acid chains on the binding partner(s). Galectins contain a carbohydrate-recognition domain (CRD) as a binding motif to recognise the glycosylation sites on other proteins and for binding. The CRD mainly detects glycoproteins and is required for post-translational modification. Their immunological roles have been established and reviewed
[29]. Different galectins have been identified that bind with immune cells, such as cytotoxic T cells, dendritic cells, and macrophages, to regulate cancer cell immunosurveillance (also listed in
Table 2). The roles of these human galectins are discussed in further detail below.
Figure 1. Locations of galectin genes in the human genome. Twenty galectin genes have been identified and are located on chromosomes 1, 11, 14, 17, 19, and 22. The partial q arms of chromosomes 1 and 11 are only shown to reflect the scale, and the scale bar represents 15 megabase pairs (Mbp) in gene distance.
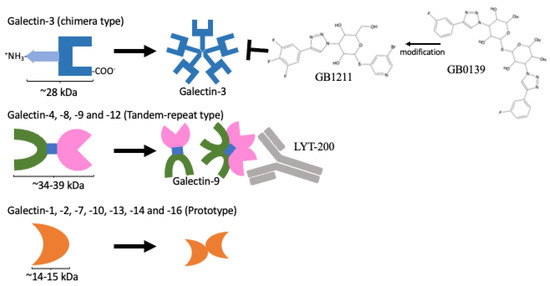
Figure 2. Protein domain structures of galectins and their inhibitors. Only the galectin-3 protein contains CRD domain and an extra amino domain allowing it to form an oligomer (
upper panel). A protein containing two distinct CRDs, galectin-4, 6, 8, 9, and 12 (
middle panel). A single CRD protein that can form homo-dimers, galectin-1, 2, 5, 7, 10, 11, 13, 14, and 16 (
lower panel). GB1211 and LYT-200 represent a newly developed galectin-3-specific inhibitor
[15] and a humanised monoclonal antibody against galectin-9, respectively, and are currently under clinical trial.
Table 2.
Intracellular and extracellular binding partners of 12 human galectins.
|
Gene/Protein Name
(Chromosome Position [30][48])
|
Intracellular
(Cytoplasmic/Nucleus)
|
Extracellular
|
|
LGALS1/Galectin-1
(Chr. 22q13.1)
|
OTX008
|
Cytoplasmic:
GRP78 [31][49]
Gemin4 [32][37]
H-Ras [33][50]
PCDH24 [34][38]
|
I
|
CC and CXC chemokines [35][42]
CD43 [36][37][51,52]
CD45 [36][38][51,53]
NRP1 [39][
Solid tumours
|
54
NCT01724320
Status unknown
(updated: 2012)
|
] |
| VEGFR2 [ 40 ] [55]
|
Galectin-3
|
Belapectin
(GR-MD-02)
|
|
LGALS2/Galectin-2
(Chr. 22q13.1)
|
| I
|
Metastatic melanoma
|
NCT02117362
Completed (updated: 2019)
|
| |
| Binds to surface of CD14(interm.–high) monocyte and promote M1 macrophage differentiation [41][56]
|
I
|
|
LGALS3/Galectin-3
(Chr. 14q22.3)
|
Metastatic melanoma, NSCLC, HNSCC
|
Cytoplasmic:
Alix (EGFR trafficking) [42][43][44][34,35,57]
Gemin4 [32][37]
K-Ras [45][46][32,40]
PCDH24 [34][38]
Nucleus:
hnRNPA2B1 [47][58]
Sp1 [48][39]
|
NCT02575404 [21]
Active (updated: 2022)
|
|
| CC and CXC chemokines | [35][42]
CD29 [49][43]
CD43 [49][43]
CD45 [49][43]
CD71 [49][43]
EGFR [50][59]
Interferon-γ [51][60]
Integrin αvβ3 [46][40]
LAG3 [52][61]
MUC1 [53][62]
|
GB1211
|
|
LGALS4/Galectin-4
(Chr. 19q13.2)
|
I
|
Healthy subjects
|
|
|
CD3 [54][63]
NCT03809052 [16]
Completed (updated: 2021)
|
|
I/II
|
|
LGALS7/Galectin-7
(Chr. 19q13.2)
|
NSCLC
|
Cytoplasmic:
Bcl-2 [55][64]
|
NCT05240131
Recruiting (updated: 2023)
|
| |
GCS-100
|
I/II
|
|
LGALS8/Galectin-8
(Chr. 1q43) |
| Relapsed/Refractory diffuse large-B-cell lymphoma
|
|
|
αM (CD11b, neutrophils) [56][65]
CD166 [57][66]
Podoplanin [58][59][67,68]
NCT00776802
Withdrawn as funding issue
(updated: 2013)
|
|
GM-CT-01
|
|
LGALS9/Galectin-9
(Chr. 17q11.2)
|
I
|
Cytoplasmic:
Binding to intracellular TIM-3 to modulate mTOR phosphorylation [60][69]
Cytoplasmic–Lysosomes:
Interact with Lamp2 to regulate lysosomal functions and autophagy [61][70] |
Breast, colorectal, head and neck, lung, prostate
|
|
4-1BB [62][71]
CD40 [63][72]
CD44 [64][73]
CD206 [65][74]
Dectin-1 (macrophages) [66][75]
DR3 [67][76]
PD-1 [68][77]
PDI [69][70][78,79]
TCR [71][72][80,81]
TIM-3 [68][73][74][77,82,83]
VISTA [75][84
NCT00054977
Completed (updated: 2012)
|
] |
|
PectaSol-C, modified
citrus pectin (MCP) [22]
|
N/A
|
Non-cancer-related: study for high blood pressure control
|
|
LGALS10/Galectin-10/Charcot-Leyden crystal protein CLC
(Chr. 19q13.2)
|
Cytoplasmic–Granules:
Eosinophil-derived neurotoxin EDN (RNS2) and eosinophil cationic protein ECP (RNS3) co-localised with CD63. It is required for the maturation of eosinophil during granulogenesis [76][85]
|
| NCT01960946
Completed (updated: 2021)
|
| |
Galactomannan/
ProLectin-M [23 |
|
LGALS12/Galectin-12/GRIP1
(Chr. 11q12.3)
|
Cytoplasmic–Endosome/Lysosomes:
VPS13C in lipid droplets and promotes the polarisation to M1 macrophage via TLR4 pathway [77][78][86,87]
|
|
|
LGALS13/Galectin-13/
placental protein 13
(Chr. 19q13.2)
|
Nucleus:
HOXA1 [79][88]
|
Binds to T lymphocytes and induces apoptosis [80][89];
Binds to neutrophils and shifts to immunoregulatory phenotype and promotes high PD-L1 expression [81][90]
|
|
LGALS14/Galectin-14
(Chr. 19q13.2)
|
|
Binds to T lymphocytes and induces apoptosis [80][89]
c-Rel [82][91]
|
|
LGALS16/Galectin-16
(Chr. 19q13.2)
|
|
c-Rel [83][92]
|
Remarks: Colour code is based on the galectin’s structure: chimera type (galectin-3) is highlighted in light blue; tandem-repeat type is highlighted in light green; prototype has not been highlighted.
3. From Bench to Bedside
3.1. Availability of Galectin-Specific Inhibitors
Luckily, due to the small size of galectin proteins, their protein structures and binding site properties have been revealed by numerous nuclear magnetic resonance (NMR) studies
[84][156]. An understanding of protein structures and functions can expedite the drug development process. Galectin inhibitors can be developed in different forms, including (1) small-molecule carbohydrates, (2) natural polysaccharides and their derivatives, (3) peptides and peptidomimetics, and (4) humanised monoclonal antibodies
[85][157]. Their structures and binding affinities to galectin-1, galectin-3, and galectin-9 were fully reviewed by Mariño et al. (2023)
[14]. Interestingly, further NMR spectroscopic analyses and competitive ELISA binding assays for eluting protein structure and substrate specifics using synthetic hydroxypropyl methacrylamide (HPMA) copolymers with multi-Galβ4GlcNAc (LacNAc), which is specific to the CRD of human galectin-1 and galectin-3 with differential affinity at the sub-nanomolar level, have been explored
[86][158]. This kind of assays might allow the identification of potential small-molecular inhibitors for future applications. In addition, peptide inhibitors and monoclonal antibodies are also useful to target secreted galectins and not interfere with the intracellular roles of galectins to minimise the side effects. New development techniques for synthesising glycosyl-side chain molecules and amino acid polypeptides may provide further new targets for galectin-based treatment in future.
3.2. Applications, Safety/Pitfalls/Limitations, and Ongoing Clinical Trials
At present, all ICIs come in the form of antibodies. This creates a limitation for future treatment potential, especially for brain metastasis, where the antibodies are unable to pass through the blood–brain barrier. In addition, the limited availability of immune cells within the central nervous system also imposes a limitation on the use of ICIs in patients with metastases at sites such as the brain and spine. The development of different small-molecule inhibitors for PD-1/PD-L1 is ongoing
[87][159]. Further understanding of the resident and infiltrating immune cells across to the brain/spine via the blood–brain barrier
[88][160] might make the impossible possible.
T-cell exhaustion is a common phenomenon in treatment using ICIs. Galectin inhibitors are a new approach to improve this situation and the durability of the treatment regimen. Combinations of ICIs and other inhibitors in the form of inhibitory antibodies or TKIs have been the subject of ongoing studies
[89][161]. All the components, including cytotoxic T cells, NK cells, and their inhibitory molecules, within the TME are required to orchestrate the anti-tumorigenic mode of killing tumour cells.
The potential pitfalls of galectin inhibitors in future treatments should be noted in drug development. The first limitation is the unknown factors associated with laboratory settings, such as the differences between mouse models and real humans. The blood components of laboratory mice are different to those of humans. Mice have a high percentage of lymphocytes (around 70% of all leukocytes). In human bodies, the predominant immune cells are neutrophils, which account for over 50% of all leukocytes
[90][162]. This issue is not easily addressed even in humanised mouse models
[91][163]. It is difficult to understand a drug or biological reagent’s effect without human trial studies. In addition, as discussed above, most galectins can alter immune responses. For example, galectin-3 has been shown to play important roles against pathogens
[92][41]. For subjects treated with galectin-3 inhibitors, infection-related adverse effects in subsequent phase 2 or 3 trial studies should be noted as a safety parameter. Treatment using galectin-9 inhibitors also raises similar concerns to galectin-3-inhibitor treatment and can affect different types of immune cells. It may also raise additional concerns regarding autoimmune issues.
Although immune-related adverse events (irAEs) are inevitable in I-O treatment, the occurrence of irAEs during treatment reflects better prognosis and overall outcomes
[93][164]. If the irAEs can be better managed and controlled using corticosteroids, this is a good sign of successful treatment. The effective and safe dosages for combined treatments could be evaluated and determined in clinical trial settings and provide guidelines for regimens and any dosage reductions in the case of irAEs. While clinical trials are still ongoing with many unforeseen hurdles to overcome, galectin-3 small-molecule inhibitors, such as belapectin and GB1211, and galectin-9 humanised monoclonal antibodies still provide new opportunities for combination therapies to tackle the current unmet and unresolved issues associated with ICI treatments for many difficult-to-treat cancers.