Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xiaodong Li and Version 2 by Catherine Yang.
The circadian clock regulates daily changes in behavioral, endocrine, and metabolic activities in mammals. Circadian rhythms in cellular physiology are significantly affected by aging. Aging induces changes in gene expression levels and rhythms in peripheral and probably central tissues.
- circadian rhythms
- peripheral clock
- inflammatory response
1. Introduction
Intrinsic circadian oscillations are present in certain nuclei of the central nervous system (e.g., the suprachiasmatic nucleus (SCN)) and most peripheral tissues in mammals [1]. At the molecular level, circadian oscillations are driven by core clock proteins that serve as transcription factors (TFs). In particular, the clock proteins CLOCK and BMAL1 dimerize to transcriptionally activate other core clock genes, such as Per1/2 and Cry1/2, whose protein products antagonize the actions of CLOCK/BMAL1, forming a negative feedback loop [1]. CLOCK/BMAL1 also activates the transcription of clock genes Nr1d1/2, whose protein products inhibit Bmal1 transcription, forming another feedback loop [1]. In addition to regulating their own transcription to sustain circadian oscillation, clock proteins also exert broad control over other genes. As a result, about 10% of genes expressed in peripheral tissues are rhythmic, and the rhythmic genes are involved in nearly all aspects of cellular functions (e.g., metabolism, immune defense, and cell cycle regulation) [2][3][2,3]. Clock functions are tissue-specific, and gene expression rhythms vary greatly across tissues. Indeed, the genomic binding of clock proteins is tissue-specific [4][5][4,5]. Those sites are found within open chromatin regions established by tissue-specific TFs (e.g., pioneer TFs) and some ubiquitously expressed TFs [6].
Core clock genes typically harbor multiple cis-elements for clock proteins themselves, a strong control thought to ensure robust and resilient clock oscillation across various cell types [6]. However, circadian rhythms are not regulated by the intrinsic tissue clock alone. Extrinsic cues, including those derived from the SCN (neural, humoral, and behavioral cues such as body temperature) and communicating signals from other tissues, also regulate gene expression rhythms [7]. Previous studies emphasize the influence of extrinsic cues on intrinsic clocks per se. Indeed, extrinsic cues can engage TFs (e.g., CREB and SRF) that regulate the transcription of the clock genes [1]. Given that TFs typically have many genomic binding sites, they could also influence other genes besides clock genes. Indeed, in the absence of rhythmic cues from the SCN and other tissues, the intrinsic clock can only sustain the rhythms of a limited set of genes (e.g., core clock genes and some clock-controlled genes). Those results indicate that clock proteins often collaborate with other TFs in rhythm regulation [6]. The collaboration can occur within the same enhancer co-bound by clock proteins and other TFs; it can also be achieved through the looping of distinct enhancers bound by clock proteins and other TFs, respectively, to promoters of their common target genes. Such combinatorial control permits plasticity in circadian rhythm regulation by allowing TFs other than clock proteins to control rhythms [6]. For example, a high-fat diet reprograms liver gene expression via PPARγ and SREBP but leaves the liver clock intact [8][9][8,9]. Lung adenocarcinoma in young mice releases cytokines to reprogram liver gene expression without disturbing the liver clock [10]. Interestingly, aging also leads to extensive reprogramming of daily gene expression in peripheral tissues of mice and man, while clock oscillations remain normal therein [11][12][13][14][11,12,13,14].
2. Mitochondrial Functions Are Rhythmic and Regulated by the Circadian Clock in Young Mice
Except for the 13 OXPHOS subunits encoded in mtDNA, all other mitochondrial proteins, including those involved in mtDNA replication, transcription, and mitochondrial protein translation, are encoded by the nuclear genome [15][75]. Some of those genes appear under clock control [16][17][76,77]. The daily changes in OXPHOS protein composition and other mitochondrial rhythms (see below) are probably optimized for daily changes in nutrient supply and energy demand associated with the feeding-fasting cycle. Depending on the feeding state, mitochondria switch fuel choice between pyruvate and fatty acid [18][79]. That mitochondrial energetics and fuel usage vary over the day is clearly evidenced by daily changes in oxygen consumption rate and the respiratory exchange ratio in mice and men [19][20][21][78,80,81]. Mitochondrial respiratory activities in vitro also change across the day, but the peak phases differ by substrates, consistent with daily changes in fuel usage [19][22][78,82]. Some mechanisms of mitochondrial fuel selection are known. For example, reversible phosphorylation of pyruvate dehydrogenase (PDH) controls mitochondrial pyruvate use. PDH is inactivated upon phosphorylation by PDH kinases and re-activated by a Ca2+-activated phosphatase [23][83]. Ca2+ influx promotes pyruvate oxidation [23][83] and is limited by MICU1 (regulating Ca2+ influx via MCU) and OPA1 present at the cristae junction [24][84]. Phosphorylation of PDH-E1α in the livers of young mice is increased during daytime, indicating that mitochondrial pyruvate oxidation is reduced during fasting [25][16]. Mitochondria undergo structural changes in response to nutrient availability [26][85]. Under nutrient-poor situations, OPA1 promotes the fusion of the mitochondrial inner membrane and intracristal assembly of OXPHOS complexes and supercomplexes to facilitate ATP production [27][28][86,87]. It was found daily changes in mitochondrial OPA1 abundance in mouse liver, with higher levels at daytime [25][16], consistent with the role of OPA1 in promoting FAO [29][88]. OPA1 not only promotes FAO but also curtails oxidative stress [30][89]. ROS production rate by mitochondria in vitro is low during FAO [31][90]. Mitochondrial oxidative stress in vivo, judged by the degree of PRDX3 dimerization in young mouse liver, is decreased during daytime, with a nadir at ZT10 [25][16]. From a metabolic point of view, mitochondrial dehydrogenases and respiratory complexes are differentially employed when different respiratory substrates are used, so the ROS production rate would vary by substrate, as indeed observed in vitro [31][90]. Low ROS production rate during FAO could be accounted for by several mechanisms. First of all, FAO produces NADH and FADH2 at an equal molar ratio. Compared to NADH, FADH2 derived from FAO donates its electrons (via ETF) to the ETC downstream of ROS production sites in complex I, thus favoring less ROS production [32][91]. Other mechanisms exist to restrain oxidative stress during FAO. For example, during FAO in the liver, TCA cycle metabolites are depleted by gluconeogenesis, and acetyl-CoAs from FAO are diverted to ketogenesis. As a result, less FADH2, NADH, and ROS are made by the TCA cycle in the liver during FAO. Moreover, a limited NADH supply to the ETC during FAO would raise the NAD+/NADH ratio, thus activating sirtuins [33][92]. Notably, the mitochondrial sirtuin SIRT3 plays a role in reducing oxidative stress. SIRT3 deacetylates various proteins [32][91], such as ETC proteins, FAO, and antioxidant defense enzymes (e.g., IDH2 and SOD2). SIRT3 increases the efficiency of ETC electron transfer to reduce ROS production and also promotes antioxidant defense [32][91]. Acetylation levels of SIRT3 targets change over the day, reaching the lowest levels near ZT10, concurrent with the nadir of mitochondrial oxidative stress in the livers of young mice [25][16]. Overall, the daily rhythm in mitochondrial redox states is closely integrated with daily metabolic changes in mouse liver, such that low oxidative stress level is associated with FAO during fasting. Consistent with clock control of mitochondrial functions [34][93], most mitochondrial rhythms in the livers of young mice, including the redox rhythm, are disrupted by the Clock△19 mutation [25][16]. The circadian clock also controls a redox rhythm at the whole cell level [35][94].3. Mitochondrial Rhythms in Mouse Liver Are Disrupted by Aging despite Normal Circadian Clock Oscillation
Intriguingly, the mitochondrial rhythms evident in young mice are disrupted by aging, due mainly to rhythm damping by age-related changes during the daytime, especially at ZT10 [25][16]. For example, mtDNA transcripts in the livers of old mice are much less abundant at ZT10, the peak time in young mice. Age-related decrease in mtDNA transcripts is also seen in mouse muscle, where nuclear-encoded OXPHOS transcripts are less affected by aging [36][95]. Changes in OXPHOS subunit composition probably lead to inefficient electron transfer and energy production and increased ROS production in various mouse tissues. Meanwhile, SIRT3 target acetylation in the livers of old mice is increased at ZT10, the nadir of corresponding rhythms in young mice. Aging abolishes the mitochondrial redox rhythm, owing to a prominent increase of oxidative stress in the livers of old mice at ZT10, the nadir of PRDX3 dimerization in young mice. The age-related disruption of mitochondrial rhythms, however, is not associated with overt clock defects. Clock gene rhythms remain normal with age in mouse liver [25][16], consistent with other studies on mouse peripheral tissues [11][12][13][11,12,13] and the SCN [37][38][39][96,97,98]. Those results indicate that aging disrupts mitochondrial rhythms through molecular mechanism(s) downstream of the circadian clock. Indeed, deep sequencing studies revealed that, while robust clock oscillations are preserved in peripheral tissues, aging induces extensive changes in gene levels and rhythms therein [11][12][13][11,12,13]. Age-related global changes in SCN gene expression await future studies. Maintaining proper circadian rhythms at the organismal and cellular levels is beneficial to health. For example, time-restricted feeding (tRF) enables robust daily rhythms to prevent metabolic diseases in young mice and men [40][99]. Age-related changes in gene expression clearly disturb metabolic processes, as evidenced by decreased rhythm amplitudes of many metabolic genes in the livers of old mice [13]. Such changes can be ameliorated by caloric restriction (CR), and the tRF factor contributes to the lifespan-extending effect of CR in mice [13]. The extent to which mitochondrial rhythms are affected by CR during aging remains to be determined.4. Mechanism(s) for Age-Related Dysregulation of Mitochondrial Rhythms
The mechanisms for age-related reprogramming of daily gene expression are under current investigation (Figure 1). Gene expression is known to be controlled by both the circadian clock and other TFs, either independently or in combination [6]. Conceivably, some TFs whose activities are altered by aging take part in reprogramming daily gene expression. Plausible candidates are TFs (e.g., NF-kB, IRFs, and STATs) activated by chronic inflammation, a hallmark of aging [41][15]. Indeed, inflammatory response genes are enriched in the tissues of old mice [11][13][11,13]. Unlike the strong immune response that disrupts the molecular clock after an LPS challenge [42][100], age-related sterile inflammation is of low grade and thus may not significantly disturb the molecular clock [43][101], at least at the early stage of aging examined [11][12][13][25][11,12,13,16].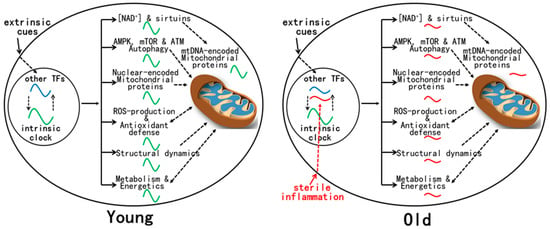
Figure 1. Mitochondrial functions are regulated by the circadian clock and other TFs in young animals. Such combinatorial regulation coordinates robust daily changes in mitochondrial protein composition, structural dynamics, energetics, and metabolism. There is a clear mitochondrial redox rhythm that is integrated with daily rhythms in metabolism. During aging, age-related activation of certain TFs, such as inflammatory TFs, leads to the reprogramming of daily gene expression that has profound effects on mitochondrial physiology, leading to the disruption of most mitochondrial rhythms that are evident in young animals. Such dysregulation of mitochondrial functions is associated with metabolic changes and disturbed redox homeostasis.