Manganese oxides are considered an essential component of natural geochemical barriers due to their redox and sorptive reactivity towards essential and potentially toxic trace elements. Despite the perception that they are in a relatively stable phase, microorganisms can actively alter the prevailing conditions in their microenvironment and initiate the dissolution of minerals, a process that is governed by various direct (enzymatic) or indirect mechanisms. Microorganisms are also capable of precipitating the bioavailable manganese ions via redox transformations into biogenic minerals, including manganese oxides (e.g., low-crystalline birnessite) or oxalates. Microbially mediated transformation influences the (bio)geochemistry of manganese and also the environmental chemistry of elements intimately associated with its oxides. Therefore, the biodeterioration of manganese-bearing phases and the subsequent biologically induced precipitation of new biogenic minerals may inevitably and severely impact the environment.
1. Introduction
Geochemical barriers are epigenetic zones with diverse functional characteristics related to their distinct physical or chemical gradients in the soil or sediment environments
[1]. They can decrease the migration capacity of chemical compounds, and, consequently, due to the accumulation of elements within their bodies, natural ore deposits are formed at these zones
[2].
One of the prevailing and vital components of the geochemical barriers are manganese oxides
[3], which affect the immobilization of both inorganic and organic compounds due to their significant sorption and redox properties
[4][5][6][4,5,6]. They are considered the strongest naturally occurring oxidants
[7], and their diverse crystalline forms serve as a pool of essential elements, including manganese. Therefore, their composition and other chemical features may affect the proper functioning of cellular metabolic pathways and the organisms’ physiological state within the geochemical barriers
[8][9][8,9].
Throughout the history of Earth’s ore formation and mineral diversification, microorganisms have been a driving force in major geological events. For example, the Great Oxidation Event (~2.2 to 2.0 Ga) and the evolution of eukaryotic microorganisms led to direct and indirect biotransformation of an initial ~1500 mineral species, resulting in an increase to over 4000 species
[10][11][10,11]. Therefore, throughout Earth’s history, autochthonous microorganisms have developed various strategies to transform and acquire manganese and other elements associated with manganese oxides
[12][13][12,13].
This is usually promoted by the interaction of reactive microbial extracellular metabolites with the surfaces of manganese phases, resulting in the gradual dissolution and transformation of its oxides
[14]. These processes are primarily mediated by the redox and protolytic reactions, in which the microorganisms (both bacteria and fungi) may be involved directly or indirectly
[15][16][15,16]. Consequently, microorganisms possess an exceptional ability to deteriorate and transform the manganese-bearing minerals, thus, altering their reactivity and stability in the environment
[17][18][19][17,18,19]. However, the biodeterioration of manganese oxides in natural geochemical barriers may also contribute to releasing associated potentially hazardous elements
[3][18][19][20][3,18,19,20], adversely affecting the environment’s vitality
[21].
In addition to the release of hazardous elements, microbially induced biodeterioration of manganese phases supports the development of sustainable agriculture by increasing the bioavailability of various essential nutrients (e.g., phosphorus and nitrogen)
[22][23][22,23] and bioavailable manganese, which plays a crucial role in various metabolic processes in plants including ROS scavenging and photosynthesis
[12][24][25][12,24,25].
From a holistic point of view, the role of microorganisms in optimal or excessive manganese availability for plants should not be overlooked. Manganese deficiency can occur in dry calcareous soils, while its toxicity occurs in poorly drained acidic soils
[26][27][26,27].
2. Manganese in Soils
Manganese can exist in different forms in soils and sediments, including Mn(II), Mn(III), and Mn(IV). This variety of oxidation states leads to numerous manganese minerals in these environments. Post
[28] reported that at least thirty different crystal structures of manganese oxides occur in the environment, with the most prevalent being birnessite, vernadite, hollandite, lithiophorite, pyrolusite, todorokite, cryptomelane, hausmannite, and romanechite. Of these, the vernadite and birnessite are the most widespread
[29], although the birnessite content can be actually lower than reported in favor of vernadite
[30].
In the soil environment, the manganese can be found in soil solution in dissolved form (mostly as complexed Mn(II)); it is adsorbed onto the surfaces of the soil mineral components and soil organic matter or sequestered in organisms. Still, the major pool of soil manganese comprises primary or secondary minerals
[31].
The manganese content in the surface soil horizons is very variable. Bowen
[32] estimated that the global average manganese concentration is 1000 mg·kg
−1, ranging from 20 mg·kg
−1 to 10,000 mg·kg
−1. The reported value is identical to the concentration of manganese in the lithosphere, which indicates the dependence of the soil manganese on its content in the parent rock
[33]. It also suggests that the manganese remains largely immobile during regional metamorphism. Thus, rock-forming minerals are the primary source of manganese in soils. There, manganese is predominantly associated with ferromagnetic silicates since Mn(II) is capable of an isomorphic substitution with Fe(II)
[34].
In sediments, the manganese is more prevalent in fine-grained fractions and primarily associates with the layered silicates, sesquioxides, and carbonates, while its content in mature quartzose sandstones is low. Therefore, the soils that are formed from mafic volcanic rocks (with a manganese content over 1000 mg·kg
−1) or shales rich in iron and magnesium contain higher amounts of manganese in comparison to soils developed from granite or sandstone (up to 400 mg·kg
−1 Mn)
[35].
Manganese is usually bound to minerals in rocks that form under reducing conditions, which predominantly causes Mn(II) to occur in the primary minerals. However, in the weathering zone, the rocks are exposed to water and permanent or fluctuating oxidizing conditions, which allows the incorporation of manganese into the weathering products. Under these conditions, it is oxidized to metastable Mn(III) or to stable Mn(IV), while Mn(II) ion is being leached out by the reactive components from the aqueous solution. From the mentioned naturally occurring manganese species, the metastable Mn(III) drives oxidative activity in organic soil layers
[36]. At the same time, the released Mn(II) ion can precipitate to form secondary minerals, e.g., oxides and oxyhydroxides
[37], and potentially can form coatings on rock surfaces and mineral particles
[28]. Therefore, the redox conditions are one of the dominating factors which control the Mn speciation in the soils. In addition to redox conditions of the soil environment, the prevailing pH is also a determining factor in manganese speciation. Under acidic soil conditions (pH < 5.5) the bioavailable Mn(II) is favored
[38], while at a higher pH range, the species of Mn(III) and Mn(IV) are likely. The increase in one pH unit leads to a 100-fold decrease in Mn(II) concentrations
[39]. Despite the natural mobilization of manganese (excluding anthropogenic sources), the concentration of its soluble forms in the surface waters only exceeds 1000 µg·L
−1 in exceptional conditions and usually does not reach the concentration of 200 µg·L
−1 [40].
The processes of manganese oxides and oxyhydroxides dissolution and precipitation regulate the mobility of manganese in soils and sediments and its availability to organisms. As
rwe
searchers mentioned earlier, the solubility of manganese oxides is primarily a function of pH. It decreases in the order of pyrochroite > hausmannite > bixbyite > manganite > birnessite > nsutite > pyrolusite (
Table 1). The dissolution of manganese oxides can also be facilitated by the presence of various chelating ligands
[41].
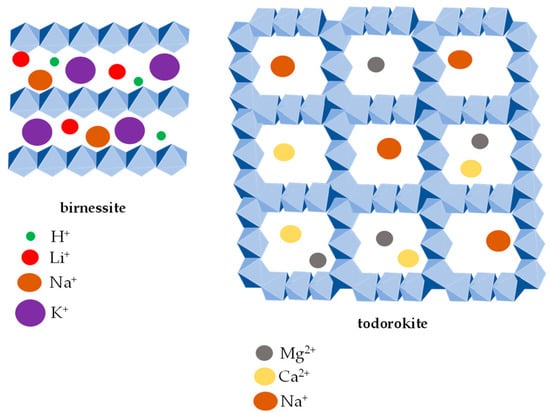
The stability of manganese oxides in the soil environment depends not only on the redox reactions and pH but also on the crystal structure of the respective mineral. The main types of crystal structures of manganese oxides include tunnel and layered structures
[28] whose elementary unit is the MnO
6 octahedron
[3]. The tunnel structure is formed by the MnO
6 octahedron chains sharing the corners of the neighboring chains, resulting in typical square or rectangular “tunnels” (
Figure 1).
Figure 1. The tunnel- and layer-type crystal structures of Mn oxides. The layer-type crystal structure is represented by birnessite (left image) with various interlayer cations H+, Li+, Na+ and K+. Todorokite (right image) represents the tunnel type (3 × 3) crystal structure with Mg2+, Ca2+ and Na+ cations in the central tunnels.
Manganese oxides with the layered structure consist of MnO
6 octahedron-based layers, and birnessite (
Figure 1) is a typical representative. Depending on the degree of hydration and the cations’ size, the layered structures may expand or collapse. The typical dimensions of the interlayer spaces are 7 or 10 Å. While the Ca(II), Mg(II), Ni(II) and Cu(II) cations stabilize the structure of 10 Å phyllomanganates, the H(I), K(I), Pb(II), Ce(III) and Th(IV) cations stimulate the collapse of the crystal structure
[48]. However, manganese with a tunnel structure (e.g., todorokite) (
Figure 1) does not collapse or expand
[49].
The layered structures of manganese oxides are the precursors of tunnel-structured oxides
[50]. The conversion between these is possible in soils, and it depends on the temperature
[51], the ratio of Mn(III) to Mn(IV)
[52], pH
[53], light conditions
[54] and the nature of the present cation between the layers of the precursors. For example, Mg(II) saturated precursors transform into todorokite
[55] while Na(I) saturated precursors convert to synthetic cryptomelane
[56].