Peritoneal carcinomatosis is a challenging condition that affects many cancer patients, and conventional therapies have limited efficacy in treating it. However, recent advances in the field of immunotherapy have shown promise in improving treatment outcomes. One promising approach is immune checkpoint inhibitors, which block proteins that inhibit T-cell activity and promote an anti-tumor immune response. Another approach involves the use of CAR-T cells, which are genetically modified T cells engineered to recognize and target cancer cells expressing specific antigens. In addition, dendritic cells and vaccine-based therapeutics are also designed to stimulate the immune system to recognize and attack cancer cells.
- intraperitoneal immunotherapy
- peritoneal carcinomatosis
- ascites
1. Introduction
2. Peritoneal Carcinomatosis
2.1. Peritoneum and Peritoneal Carcinomatosis
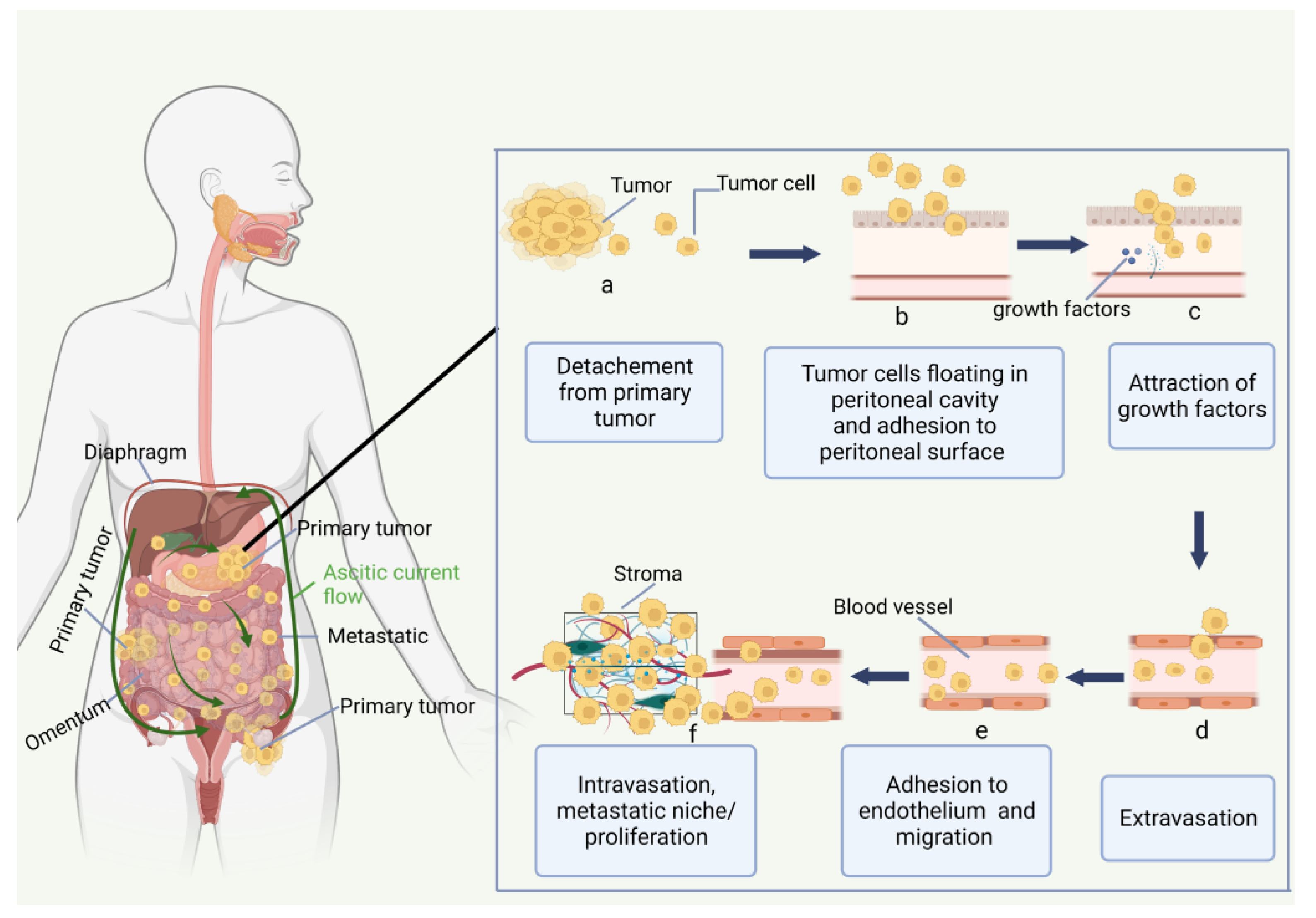
2.2. Immune Environment of Peritoneal Carcinomatosis
The innate (neutrophils, macrophages, dendritic cells, and natural killer cells) and the adaptive (B and T lymphocytes) immune systems can recognize and destroy tumor cells. However, cancer cells gain the ability to evade immune surveillance by targeting or manipulating the immune system. Since lymph nodes and the greater omentum both include immune cells such as macrophages and lymphocytes, immune cell activation may be a potential PC treatment strategy [16][22]. The peritoneal cavity has immunologically competent cells, such as 45% of monocytes/macrophages (CD68+), 45% of T-lymphocytes (CD2+), 8% of NK-cells (natural killer cells), and 2% of dendritic cells, as well as A substantial proportion of CD4+ (92%) and CD8+ (73%). Approximately 49% of the cells in the peritoneum were positive for class II major histocompatibility complex antigens [26][23]. In contrast to CD45RO-naive T-lymphocytes, CD45RO+ T-lymphocytes have already differentiated into memory and effector T cells. In contrast to peripheral blood cells, which have a predominance of CD8+ T-lymphocytes, healthy individuals have an inverted CD4+/CD8+ T-lymphocyte ratio. Innate immunity is activated by mesenchymal precursors of the peritoneum. Interleukin-1 (IL-1), interleukin-6 (IL-6), prostaglandin E2, granulocyte stimulating factor (GCSF), granulocyte monocyte colony-stimulating factor (GM-CSF), monocyte colony-stimulating factor (MCSF), and vascular epithelial growth factor (VEGF) are all pro-inflammatory mediators that are released by mesenchymal cells. Figure 2 shows the cell ascites tumor microenvironment (TME).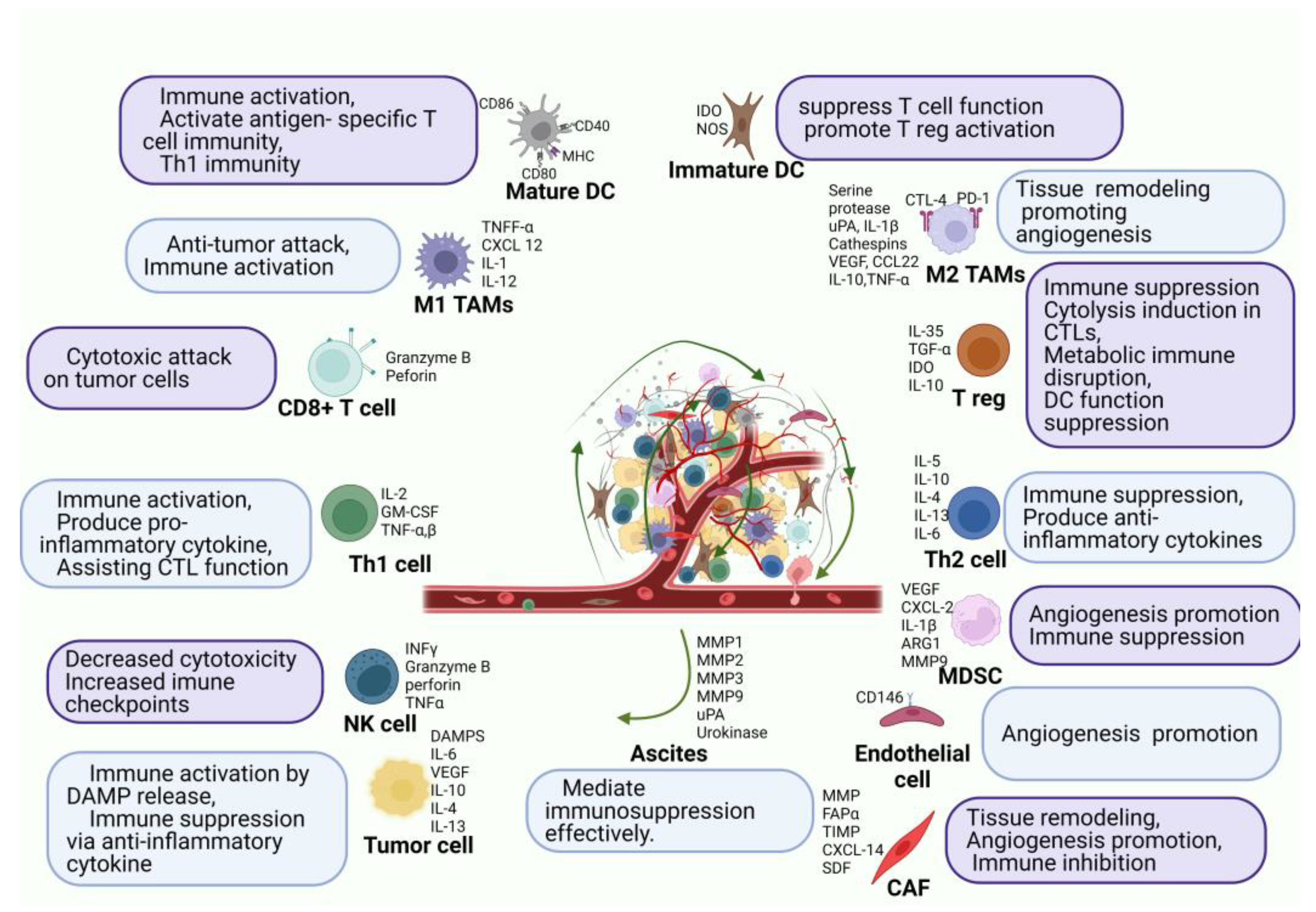
3. Immune Checkpoint Inhibitors
Patients with a wide variety of cancers benefit from antibodies that target immunological checkpoints, such as cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), PD-1, and PD-L1 [37][30]. In 2011, for the first time, the FDA approved ipilimumab as an immune checkpoint inhibitor (ICI) for the treatment of metastatic melanoma [38][31]. ICI has shown excellent and, most importantly, long-lasting responses in advanced tumor patients, unlike targeted treatment and chemotherapy. It is known that factors such as microsatellite instability (MSI-H), tumor mutation burden (TMB), and PD-L1 expression can be used to predict the therapeutic response of an ICI [41][32]. However, there is still insufficient evidence on whether these factors have a therapeutic effect in peritoneal metastasis. MSI-H cancers occur in gastrointestinal (colorectal, gastric, hepato-biliary) and endometrial malignancies and are caused by germline mutations in one of the DNA mismatch repair genes or somatic promoter hypermethylation of MLH. A high tumor mutation load boosts immunogenicity and ICI sensitivity [42][33]. After establishing a genetic signature-based predictive biomarker for systemic therapy (pembrolizumab, anti-PD-1 ICI across many tumor types), the FDA awarded its first tissue-agnostic clearance for deficient mismatch repair (dMMR)/MSI-H malignancies [43][34]. The FDA approval of pembrolizumab in dMMR/MSI-H was based on the KEYNOTE-158 study. The study evaluated the efficacy of pembrolizumab in patients with advanced solid tumors that had progressed on standard therapy. They found that pembrolizumab showed promising results in patients with dMMR/MSI-H solid tumors, with an overall response rate of 34.3% and a median duration of response of 24.4 months. The study suggests that pembrolizumab may be an effective treatment option for patients with dMMR/MSI-H solid tumors, including some pancreatic cancers [44][35]. In research utilizing a newly produced highly metastatic clone of murine gastric cancer cells, YTN16P, it was shown that infusion of PD-1 mAb through the intravenous or intraperitoneal route lowered the rate of metastasis development on the mesenteric surface by 30–40% as a monotherapy [46][36]. Additionally, previous studies utilizing colon [40,47][37][38] or ovarian cancer cells [48,49,50][39][40][41] have shown that anti-PD-1 mAb may partially, but not fully, inhibit the development of PM in immunocompetent animals. Mouse models established using YTN16 and LmcMF are resistant to ICI treatment because CXCL12 derived from CAFs recruit M2 macrophages which secrete various cytokines, such as VEGF, IL-10, amphiregulin, and MMP-1 [51][42]. These cytokines exhaust CD8+ cells, either directly or indirectly. Furthermore, infiltration of CD8+ cells is inhibited due to the high intertumoral pressure associated with tumor fibrosis induced by CAFs. Although these models are resistant to ICI therapy, anti-CAF treatment recovered the therapeutic efficacy of the ICI [52][43].4. Monoclonal Antibodies
4.1. MOC31PE Immunotoxin
The monoclonal antibody called MOC31PE is derived from the Pseudomonas exotoxin A (PE) and targets the epithelial cell adhesion molecule (EpCAM), a transmembrane glycoprotein that is significantly overexpressed in cancerous tissue, including HGSOC, and is expressed at small levels in normal tissue [59,60][44][45]. After attaching to the EpCAM-expressing surface of cancer cells, MOC31PE kills cells by deactivating crucial cellular functions. Additionally, MOC31PE has a competitive edge over earlier anti-EpCAM antibody-based treatments due to its “simpler” mode of action, requiring just binding to EpCAM-expressing cancer cells before directly promoting cancer cell death through toxin release inside the target cells [61,62][46][47]. The therapeutic efficacy of chemotherapy-resistant cancer cells can be enhanced by MOC31PE. Recently, it was shown that patients with metastatic carcinomas who express EpCAM showed good tolerance to systemic doses of MOC31PE [63][48]. The ImmunoPeCa experiment (NCT02219893), a phase 1 dose-escalation study carried out in 2017 [65][49], examined patients with peritoneal metastasis from colorectal cancer (CRC) after demonstrating anti-cancer efficacy in preclinical testing [61,64,66][46][50][51]. The MOC31PE immunotoxin was given intraperitoneally the day following surgery to 21 patients who had CRS/HIPEC for PC from CRC at four distinct dose levels. The medicine was found to be safe and well-tolerated, with no evidence of dose-limiting harm. Even though MOC31PE was not absorbed into the body very much, the levels in the peritoneal fluid were thought to be cytotoxic. Neutralizing antibodies were produced by all patients.4.2. Catumaxomab
Catumaxomab is a rat-murine bispecific and trifunctional antibody that targets EpCAM and can have a long-lasting immunization effect [70,71][52][53]. In 2009, catumaxomab was approved in Europe as the first drug for malignant ascites linked to PC [72,73][54][55]. This bispecific monoclonal antibody can target immune systems and has a safe profile in clinical trials when administered intravenously (IP). Catumaxomab’s fragment-crystallizable (Fc) domain activates Fc-receptor types I, IIa, and III on NK cells, CD3+ T-cells, and EpCAM receptors, which are the substance’s two antigen-binding sites that it particularly targets. As a result of this mechanism, pro-apoptotic cytokines including IL-2, IL-12, and TNF phagocytose the targeted tumor cells, leading to cell death [74,75,76][56][57][58]. Many ovarian cancer patients may already have peritoneal metastases at the time of their diagnosis, and the presence of a significant amount of malignant ascites accelerates the disease’s development and distention. In a trial by Burges et al., catumaxomab was used to treat ascites in 23 ovarian cancer patients who had resistant to conventional treatment. Production of ascites was significantly reduced during catumaxomab therapy in response to increasing dosages. Twenty-eight days after the last infusion, only one of twenty-three patients who received treatment needed a paracentesis, which is still nearly 2 weeks longer than is usually necessary [82,83][59][60]. In a multicenter trial conducted by Wimberger et al., 258 patients with ovarian and non-gynecologic malignancies were randomly assigned to treatment and control groups to determine the effect of catumaxomab therapy on life quality. Patient surveys were used to determine the results. Compared to paracentesis alone, treatment with catumaxomab with paracentesis considerably delayed the period until the quality of life deteriorated [84][61]. The therapy of malignant ascites from EpCAM+ tumors was evaluated in a randomized, multicenter study by Heiss et al. Catumaxomab significantly enhanced median puncture-free survival and time to next therapeutic intervention in the experimental group, as well as overall survival, among 258 patients with gastric cancer in this phase II/III clinical trial [75][57].5. Cancer Vaccines for Peritoneal Metastasis
Therapeutic vaccines against cancer are a further immunotherapy strategy that has attracted substantial recent advancements in the intraperitoneal developments of PC. Malignant ascites have a bad prognosis and are a significant barrier to the immune system responding to vaccines. To combat this, vaccines are currently being developed and modified to specifically target ascites to enhance the quality of life for PM patients.
Cellular, viral vector, and molecular (peptide, DNA, or RNA) are the three main platforms for cancer vaccines [86][62]. Allogeneic tumor cell lines or autologous patient-derived tumor cells are used to create cellular vaccines [87][63]. Due to their functions as tumor antigen consumers, processors, and presenters, dendritic cells (DCs) are employed to create cellular cancer vaccines. Oncolytic viral vaccinations have been genetically altered to target and kill tumor cells [88][64]. In addition to their oncolytic effects, viral vectors also stimulate tumor-specific immune responses by providing tumor antigens through more typical T-cell priming procedures [89][65]. On the cell surface, major histocompatibility complex (MHC) peptides expression can be detected by T-cells [90][66]. For the creation of peptide-based cancer vaccines, it is important to know how peptides and T cell receptors interact with MHC. Enzymes break down short peptides, which are typically nine amino acid residues long, and immediately connect to MHC molecules, perhaps generating tolerance [91][67]. Longer peptides, typically 30 mer, are taken in by antigen-presenting cells (APCs), processed for MHC presentation, and result in memory CD4+ and CD8+ T cell immunological responses, which may make APCs more immunogenic [91][67]. DNA vaccines, often known as “naked DNA”, are closed circular DNA plasmids that encode TAAs and immunomodulatory substances intending to induce tumor-specific immune responses [92][68]. Despite being straightforward, secure, and quick to create, naked DNA vaccines are ineffective against target tumor cells due to low rates of transfection. mRNA vaccines, which are produced in vitro, encode an antigen or antigens, and following internalization, they express proteins that cause an immune reaction. mRNA vaccines may convey a large number of antigens and co-stimulatory signals without running the risk of infection or insertional mutagenesis, and their manufacture is rapid and affordable. However, the delivery effectiveness and stability are issues for mRNA vaccines [92][68].
Targeting ascites in PC has been accomplished by combining DCs with cytokine-induced killer cells (CIKs), which are cytotoxic T lymphocytes with a CD3+ CD56+ phenotype. The choice of CIKs was made based on three important criteria: they exhibit low cytotoxicity toward normal cells, no negative impact on hematopoiesis in the bone marrow, and resistance to Fas ligand-induced apoptosis. The effects of the combined treatment of DCs and CIKs include an increase in cytotoxic T cells in ascites that are driven by TNF and IFN and a reduction in immunosuppressive Tregs [93][69]. Similar to CAR-T cells, the method by which cancer vaccines are administered plays an important role in their dissemination. Natural killer cells (NKs) and dendritic cells (DCs) working together to fight tumors have been proven to be successful. Geller et al. demonstrated that IP- injection of IL-2-activated NK cells enhanced antitumor effects in an ovarian cancer mouse model xenograft, in contrast to systemic distribution [94][70].
Many cycles of patient-derived type I CD4+ T helper cells (Th1) provided by IP together with the cytokines IL-2 and IFN were shown to improve the anti-tumor activity of autologous CD8+ T cells against the tumor-specific glycoform of MUC1. This was reported by Dobrzanski et al. [98,99][71][72]. In a peritoneal metastatic colon cancer murine model, Alkayyal et al. further emphasized the relevance of combining pro-inflammatory cytokine IL-12 with an oncolytic virus (Maraba MG1) for reducing tumor burden in a CT26 colon cancer model. When MG1-IL12-ICV was IP-administered to these animals, it significantly decreased tumor development, created resistance to CT26 cell reinoculation, and improved survival. Regarding the mechanism, IL-12 was effective in enticing NK cells to the tumor location for annihilation. When paired with MG1 viral proteins, these activated NK cells generated IFN, which stimulated DCs and aided in the attraction of more NK cells [100][73].
6. CAR-T Cell Therapy for Peritoneal Carcinomatosis
6.1. Basic of CAR-T Cells
CAR-T cells have undergone genetic alteration to express chimeric receptors that allow them to target certain surface antigens regardless of a person’s major histocompatibility class. Although Gross et al. initially described this type of modified T cell in 1989, this technology has only evolved dramatically in the last decade, particularly for the treatment of hematologic malignancies [109][74]. Immunotherapy has grown in popularity since the development of CAR-T cells, which allow T cells to produce synthetic receptors against specific surface antigens and destroy tumor cells [110][75]. These antigens may bind to carbohydrates, glycolipids, proteoglycans, and proteins [111,112][76][77]. As a result of CAR-T cells’ therapeutic success in clinical trials of hematologic malignancies, more research is being conducted on its application to the treatment of therapy-resistant stage IV solid tumors. CAR-T cells are composed of extracellular single-chain variable fragments (scFv) of antibodies specific to the target tumor antigen and the T-cell activation domain. CAR-Ts, in contrast to specialized T-cell therapy, are MHC-independent due to the scFv component [113,114][78][79].6.2. Administration Route of CAR-T for Peritoneal Carcinomas
Katz et al. provided the initial description of CAR-T therapy for PC, using CEA-targeting CAR-T cells to treat colorectal PC in an animal model. The authors noticed that intraperitoneal dispersion was superior to systemic injection. Compared to systemic therapy, intraperitoneal injection resulted in a higher tumor decrease and a longer-lasting impact. These data suggest that protection against recurrence and other distant metastases may be possible [121][80]. Ang et al. evaluated a mouse model of PC by employing mRNA transfection to generate CAR-T cells against EpCAM. This type of transfection had temporary effects, boosting safety in the case of adverse consequences. Due to the transient nature of the effect, repeated infusions are necessary for optimal outcomes [123][81]. These data suggest that local injection boosts CAR-T cell infiltration and trafficking, increases anticancer activity, increases recurrence protection, and enhances extraperitoneal antitumor efficacy while limiting systemic adverse effects. Solid tumors and PC still pose specific obstacles that must be solved. The microenvironment of the tumor, which generates an immunological and physical barrier, is the greatest impediment. The stroma of the tumor, which is rich in collagen in the extracellular matrix, is one of the components of the physical barrier. Due to the stroma, the tumor cells cannot be treated locally or systemically. However, collagenase can degrade this collagen, facilitating medication penetration [124][82].6.3. CAR-T Cell Studies for Peritoneal Carcinomatosis
The transfer of T lymphocytes with the CAR (chimeric antigen receptor) gene, which is selective for tumor-associated antigens, across regional boundaries (TAAs) to the peritoneal cavity enhances the transfer of CAR-T cells to the disease location while decreasing or eliminating neurotoxicity and cytokine release syndrome. CAR-T cells were examined as a potential new treatment option for ovarian cancer, which is often diagnosed in a late stage. Koneru et al. focused on the expanded extracellular domain MUC16 (MUC-16ecto) when treating advanced-stage ovarian cancer. To ensure that these CAR-Ts would activate and proliferate at the location of the tumor in the presence of immunological checkpoints, researchers engineered anti-MUC-16ecto CAR-T cells that produced IL-12. In a SCID Beige ovarian cancer xenograft model, these CAR-Ts had more efficacy than anti-MUC-16ecto CAR-T cells without an IL-12 arm to enhance antitumor activity and mouse survival when delivered intraperitoneally (IP) [140][83].References
- Willaims, S.C.P. Peritoneal Carcinomatosis: Cause, Symptoms, Diagnosis, and Treatment. Available online: https://www.webmd.com/cancer/what-is-peritoneal-carcinomatosis (accessed on 9 July 2022).
- Coccolini, F.; Gheza, F.; Lotti, M.; Virzì, S.; Iusco, D.; Ghermandi, C.; Melotti, R.; Baiocchi, G.; Giulini, S.M.; Ansaloni, L.; et al. Peritoneal Carcinomatosis. World J. Gastroenterol. 2013, 19, 6979–6994.
- McMullen, J.R.W.; Selleck, M.; Wall, N.R.; Senthil, M. Peritoneal Carcinomatosis: Limits of Diagnosis and the Case for Liquid Biopsy. Oncotarget 2017, 8, 43481–43490.
- Shariat-Madar, B.; Jayakrishnan, T.T.; Gamblin, T.C.; Turaga, K.K. Surgical Management of Bowel Obstruction in Patients with Peritoneal Carcinomatosis. J. Surg. Oncol. 2014, 110, 666–669.
- Glass, R.L.; LeDuc, R.J. Small Intestinal Obstruction from Peritoneal Carcinomatosis. Am. J. Surg. 1973, 125, 316–317.
- Chu, D.Z.; Lang, N.P.; Thompson, C.; Osteen, P.K.; Westbrook, K.C. Peritoneal Carcinomatosis in Nongynecologic Malignancy. A Prospective Study of Prognostic Factors. Cancer 1989, 63, 364–367.
- Kerscher, A.G.; Chua, T.C.; Gasser, M.; Maeder, U.; Kunzmann, V.; Isbert, C.; Germer, C.T.; Pelz, J.O.W. Impact of Peritoneal Carcinomatosis in the Disease History of Colorectal Cancer Management: A Longitudinal Experience of 2406 Patients over Two Decades. Br. J. Cancer 2013, 108, 1432–1439.
- Yonemura, Y.; Bandou, E.; Kinoshita, K.; Kawamura, T.; Takahashi, S.; Endou, Y.; Sasaki, T. Effective Therapy for Peritoneal Dissemination in Gastric Cancer. Surg. Oncol. Clin. N. Am. 2003, 12, 635–648.
- Thomassen, I.; Bernards, N.; van Gestel, Y.R.; Creemers, G.-J.; Jacobs, E.M.; Lemmens, V.E.; de Hingh, I.H. Chemotherapy as Palliative Treatment for Peritoneal Carcinomatosis of Gastric Origin. Acta. Oncol. 2014, 53, 429–432.
- Sugarbaker, P.H.; Jablonski, K.A. Prognostic Features of 51 Colorectal and 130 Appendiceal Cancer Patients with Peritoneal Carcinomatosis Treated by Cytoreductive Surgery and Intraperitoneal Chemotherapy. Ann. Surg. 1995, 221, 124–132.
- Mahteme, H.; Hansson, J.; Berglund, A.; Påhlman, L.; Glimelius, B.; Nygren, P.; Graf, W. Improved Survival in Patients with Peritoneal Metastases from Colorectal Cancer: A Preliminary Study. Br. J. Cancer 2004, 90, 403–407.
- Verwaal, V.; van Ruth, S.; de Bree, E.; van Sloothen, G.; van Tinteren, H.; Boot, H.; Zoetmulder, F. Randomized Trial of Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy versus Systemic Chemotherapy and Palliative Surgery in Patients with Peritoneal Carcinomatosis of Colorectal Cancer. J. Clin. Oncol. 2003, 21, 3737–3743. Available online: https://pubmed.ncbi.nlm.nih.gov/14551293/ (accessed on 14 July 2022).
- Van Oudheusden, T.R.; Nienhuijs, S.W.; Luyer, M.D.; Nieuwenhuijzen, G.A.; Lemmens, V.E.; Rutten, H.J.; de Hingh, I.H. Incidence and Treatment of Recurrent Disease after Cytoreductive Surgery and Intraperitoneal Chemotherapy for Peritoneally Metastasized Colorectal Cancer: A Systematic Review. Eur. J. Surg. Oncol. 2015, 41, 1269–1277.
- Karunasena, E.; Sham, J.; McMahon, K.W.; Ahuja, N. Genomics of Peritoneal Malignancies. Surg. Oncol. Clin. N. Am. 2018, 27, 463–475.
- Slavin, T.; Neuhausen, S.L.; Rybak, C.; Solomon, I.; Nehoray, B.; Blazer, K.; Niell-Swiller, M.; Adamson, A.W.; Yuan, Y.-C.; Yang, K.; et al. Genetic Gastric Cancer Susceptibility in the International Clinical Cancer Genomics Community Research Network. Cancer Genet. 2017, 216–217, 111–119.
- Ströhlein, M.; Heiss, M.; Jauch, K.-W. The Current Status of Immunotherapy in Peritoneal Carcinomatosis. Expert Rev. Anticancer. Ther. 2016, 16, 1019–1027.
- Kanda, M.; Kodera, Y. Molecular Mechanisms of Peritoneal Dissemination in Gastric Cancer. World J. Gastroenterol. 2016, 22, 6829–6840.
- Li, X.; Ji, Z.; Li, Y. Peritoneal Carcinomatosis Diagnosis and Treatment in China: Focusing on Training and Collaboration. Indian J. Surg. Oncol. 2019, 10, 12–18.
- Sadeghi, B.; Arvieux, C.; Glehen, O.; Beaujard, A.C.; Rivoire, M.; Baulieux, J.; Fontaumard, E.; Brachet, A.; Caillot, J.L.; Faure, J.L.; et al. Peritoneal Carcinomatosis from Non-Gynecologic Malignancies: Results of the EVOCAPE 1 Multicentric Prospective Study. Cancer 2000, 88, 358–363.
- Chia, C.S.; You, B.; Decullier, E.; Vaudoyer, D.; Lorimier, G.; Abboud, K.; Bereder, J.-M.; Arvieux, C.; Boschetti, G.; Glehen, O.; et al. Patients with Peritoneal Carcinomatosis from Gastric Cancer Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Is Cure a Possibility? Ann. Surg. Oncol. 2016, 23, 1971–1979.
- Verwaal, V.J.; Bruin, S.; Boot, H.; van Slooten, G.; van Tinteren, H. 8-Year Follow-up of Randomized Trial: Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy versus Systemic Chemotherapy in Patients with Peritoneal Carcinomatosis of Colorectal Cancer. Ann. Surg. Oncol. 2008, 15, 2426–2432.
- Yao, X.; Ajani, J.; Song, S. Molecular Biology and Immunology of Gastric Cancer Peritoneal Metastasis. Transl. Gastroenterol. Hepatol. 2020, 5, 57.
- Kubicka, U.; Olszewski, W.L.; Tarnowski, W.; Bielecki, K.; Ziółkowska, A.; Wierzbicki, Z. Normal Human Immune Peritoneal Cells: Subpopulations and Functional Characteristics. Scand. J. Immunol. 1996, 44, 157–163.
- Bagheri, V.; Abbaszadegan, M.R.; Memar, B.; Motie, M.R.; Asadi, M.; Mahmoudian, R.A.; Gholamin, M. Induction of T Cell-Mediated Immune Response by Dendritic Cells Pulsed with MRNA of Sphere-Forming Cells Isolated from Patients with Gastric Cancer. Life Sci. 2019, 219, 136–143.
- Fujimori, D.; Kinoshita, J.; Yamaguchi, T.; Nakamura, Y.; Gunjigake, K.; Ohama, T.; Sato, K.; Yamamoto, M.; Tsukamoto, T.; Nomura, S.; et al. Established Fibrous Peritoneal Metastasis in an Immunocompetent Mouse Model Similar to Clinical Immune Microenvironment of Gastric Cancer. BMC Cancer 2020, 20, 1014.
- Park, H.S.; Kwon, W.S.; Park, S.; Jo, E.; Lim, S.J.; Lee, C.-K.; Lee, J.B.; Jung, M.; Kim, H.S.; Beom, S.-H.; et al. Comprehensive Immune Profiling and Immune-Monitoring using Body Fluid of Patients with Metastatic Gastric Cancer. J. Immunother. Cancer 2019, 7, 268.
- Lim, B.; Kim, C.; Kim, J.-H.; Kwon, W.S.; Lee, W.S.; Kim, J.M.; Park, J.Y.; Kim, H.S.; Park, K.H.; Kim, T.S.; et al. Genetic Alterations and their Clinical Implications in Gastric Cancer Peritoneal Carcinomatosis Revealed by Whole-Exome Sequencing of Malignant Ascites. Oncotarget 2016, 7, 8055–8066.
- Sasada, T.; Kimura, M.; Yoshida, Y.; Kanai, M.; Takabayashi, A. CD4+CD25+ Regulatory T Cells in Patients with Gastrointestinal Malignancies: Possible Involvement of Regulatory T Cells in Disease Progression. Cancer 2003, 98, 1089–1099.
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The Contribution of Vascular Endothelial Growth Factor to the Induction of Regulatory T-Cells in Malignant Effusions. Antcancer Res. 2009, 29, 881–888.
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Chia, D.K.A.; Gwee, Y.X.; Sundar, R. Resistance to Systemic Immune Checkpoint Inhibition in the Peritoneal Niche. J. Immunother. Cancer 2022, 10, e004749.
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, PO.17.00073.
- Casak, S.J.; Marcus, L.; Fashoyin-Aje, L.; Mushti, S.L.; Cheng, J.; Shen, Y.-L.; Pierce, W.F.; Her, L.; Goldberg, K.B.; Theoret, M.R.; et al. FDA Approval Summary: Pembrolizumab for the First-Line Treatment of Patients with MSI-H/DMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021, 27, 4680–4684.
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10.
- Kumagai, Y.; Futoh, Y.; Miyato, H.; Ohzawa, H.; Yamaguchi, H.; Saito, S.; Kurashina, K.; Hosoya, Y.; Lefor, A.K.; Sata, N.; et al. Effect of Systemic or Intraperitoneal Administration of Anti-PD-1 Antibody for Peritoneal Metastases from Gastric Cancer. Vivo 2022, 36, 1126–1135.
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980.
- Lee, Y.S.; Lee, W.S.; Kim, C.W.; Lee, S.J.; Yang, H.; Kong, S.J.; Ning, J.; Yang, K.-M.; Kang, B.; Kim, W.R.; et al. Oncolytic Vaccinia Virus Reinvigorates Peritoneal Immunity and Cooperates with Immune Checkpoint Inhibitor to Suppress Peritoneal Carcinomatosis in Colon Cancer. J. Immunother. Cancer 2020, 8, e000857.
- Wei, H.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, M.; Hellstrom, I.; Hellstrom, K.E.; et al. Combinatorial PD-1 Blockade and CD137 Activation Has Therapeutic Efficacy in Murine Cancer Models and Synergizes with Cisplatin. PLoS ONE 2013, 8, e84927.
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 Blockade and OX40 Triggering Synergistically Protects against Tumor Growth in a Murine Model of Ovarian Cancer. PLoS ONE 2014, 9, e89350.
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 Blockade and GITR Triggering Induce a Potent Antitumor Immunity in Murine Cancer Models and Synergizes with Chemotherapeutic Drugs. J. Transl. Med. 2014, 12, 36.
- Yamaguchi, T.; Fushida, S.; Yamamoto, Y.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Miyashita, T.; Tajima, H.; Ninomiya, I.; Munesue, S.; et al. Tumor-Associated Macrophages of the M2 Phenotype Contribute to Progression in Gastric Cancer with Peritoneal Dissemination. Gastric Cancer 2016, 19, 1052–1065.
- Nakamura, Y.; Kinoshita, J.; Yamaguchi, T.; Aoki, T.; Saito, H.; Hamabe-Horiike, T.; Harada, S.; Nomura, S.; Inaki, N.; Fushida, S. Crosstalk between Cancer-Associated Fibroblasts and Immune Cells in Peritoneal Metastasis: Inhibition in the Migration of M2 Macrophages and Mast Cells by Tranilast. Gastric Cancer 2022, 25, 515–526.
- Bellone, S.; Siegel, E.R.; Cocco, E.; Cargnelutti, M.; Silasi, D.-A.; Azodi, M.; Schwartz, P.E.; Rutherford, T.J.; Pecorelli, S.; Santin, A.D. Overexpression of Epithelial Cell Adhesion Molecule in Primary, Metastatic, and Recurrent/Chemotherapy-Resistant Epithelial Ovarian Cancer: Implications for Epithelial Cell Adhesion Molecule-Specific Immunotherapy. Int. J. Gynecol. Cancer 2009, 19, 860–866.
- Köbel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Ionescu, D.N.; Rajput, A.; et al. Ovarian Carcinoma Subtypes are Different Diseases: Implications for Biomarker Studies. PLoS Med. 2008, 5, e232.
- Andersson, Y.; Engebraaten, O.; Fodstad, Ø. Synergistic Anti-Cancer Effects of Immunotoxin and Cyclosporin in Vitro and in Vivo. Br. J. Cancer 2009, 101, 1307–1315.
- Andersson, Y.; Juell, S.; Fodstad, Ø. Downregulation of the Antiapoptotic MCL-1 Protein and Apoptosis in MA-11 Breast Cancer Cells Induced by an Anti-Epidermal Growth Factor Receptor-Pseudomonas Exotoxin a Immunotoxin. Int. J. Cancer 2004, 112, 475–483.
- Andersson, Y.; Engebraaten, O.; Juell, S.; Aamdal, S.; Brunsvig, P.; Fodstad, Ø.; Dueland, S. Phase I Trial of EpCAM-Targeting Immunotoxin MOC31PE, Alone and in Combination with Cyclosporin. Br. J. Cancer 2015, 113, 1548–1555.
- Frøysnes, I.S.; Andersson, Y.; Larsen, S.G.; Davidson, B.; Øien, J.-M.T.; Olsen, K.H.; Giercksky, K.-E.; Julsrud, L.; Fodstad, Ø.; Dueland, S.; et al. Novel Treatment with Intraperitoneal MOC31PE Immunotoxin in Colorectal Peritoneal Metastasis: Results From the ImmunoPeCa Phase 1 Trial. Ann. Surg. Oncol. 2017, 24, 1916–1922.
- Wiiger, M.T.; Bideli, H.; Fodstad, O.; Flatmark, K.; Andersson, Y. The MOC31PE Immunotoxin Reduces Cell Migration and Induces Gene Expression and Cell Death in Ovarian Cancer Cells. J. Ovarian Res. 2014, 7, 23.
- Flatmark, K.; Guldvik, I.J.; Svensson, H.; Fleten, K.G.; Flørenes, V.A.; Reed, W.; Giercksky, K.-E.; Fodstad, Ø.; Andersson, Y. Immunotoxin Targeting EpCAM Effectively Inhibits Peritoneal Tumor Growth in Experimental Models of Mucinous Peritoneal Surface Malignancies. Int. J. Cancer 2013, 133, 1497–1506.
- Ströhlein, M.A.; Heiss, M.M. Intraperitoneal Immunotherapy to Prevent Peritoneal Carcinomatosis in Patients with Advanced Gastrointestinal Malignancies. J. Surg. Oncol. 2009, 100, 329–330.
- Jäger, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Ströhlein, M.; Theissen, B.; Heiss, M.M.; et al. Immunomonitoring Results of a Phase II/III Study of Malignant Ascites Patients Treated with the Trifunctional Antibody Catumaxomab (Anti-EpCAM × Anti-CD3). Cancer Res. 2012, 72, 24–32.
- Ströhlein, M.A.; Heiss, M.M. Immunotherapy of Peritoneal Carcinomatosis. Cancer Treat. Res. 2007, 134, 483–491.
- Chelius, D.; Ruf, P.; Gruber, P.; Plöscher, M.; Liedtke, R.; Gansberger, E.; Hess, J.; Wasiliu, M.; Lindhofer, H. Structural and Functional Characterization of the Trifunctional Antibody Catumaxomab. MAbs 2010, 2, 309–319.
- Seimetz, D. Novel Monoclonal Antibodies for Cancer Treatment: The Trifunctional Antibody Catumaxomab (Removab®). J. Cancer 2011, 2, 309–316.
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The Trifunctional Antibody Catumaxomab for the Treatment of Malignant Ascites Due to Epithelial Cancer: Results of a Prospective Randomized Phase II/III Trial. Int. J. Cancer 2010, 127, 2209–2221.
- Heiss, M.M.; Ströhlein, M.A.; Jäger, M.; Kimmig, R.; Burges, A.; Schoberth, A.; Jauch, K.-W.; Schildberg, F.-W.; Lindhofer, H. Immunotherapy of Malignant Ascites with Trifunctional Antibodies. Int. J. Cancer 2005, 117, 435–443.
- Burges, A.; Wimberger, P.; Kümper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective Relief of Malignant Ascites in Patients with Advanced Ovarian Cancer by a Trifunctional Anti-EpCAM × Anti-CD3 Antibody: A Phase I/II Study. Clin. Cancer Res. 2007, 13, 3899–3905.
- Mackey, J.R.; Venner, P.M. Malignant Ascites: Demographics, Therapeutic Efficacy and Predictors of Survival. Can. J. Oncol. 1996, 6, 474–480.
- Wimberger, P.; Gilet, H.; Gonschior, A.-K.; Heiss, M.M.; Moehler, M.; Oskay-Oezcelik, G.; Al-Batran, S.-E.; Schmalfeldt, B.; Schmittel, A.; Schulze, E.; et al. Deterioration in Quality of Life (QoL) in Patients with Malignant Ascites: Results from a Phase II/III Study Comparing Paracentesis plus Catumaxomab with Paracentesis Alone. Ann. Oncol. 2012, 23, 1979–1985.
- Hollingsworth, R.E.; Jansen, K. Turning the Corner on Therapeutic Cancer Vaccines. NPJ Vaccines 2019, 4, 7.
- Le, D.T.; Pardoll, D.M.; Jaffee, E.M. Cellular Vaccine Approaches. Cancer J. 2010, 16, 304–310.
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing Oncolytic Virotherapy in Cancer Treatment. Nat. Rev. Drug. Discov. 2019, 18, 689–706.
- Osipov, A.; Murphy, A.; Zheng, L. From Immune Checkpoints to Vaccines: The Past, Present and Future of Cancer Immunotherapy. Adv. Cancer Res. 2019, 143, 63–144.
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292.
- Slingluff, C.L. The Present and Future of Peptide Vaccines for Cancer: Single or Multiple, Long or Short, Alone or in Combination? Cancer J. 2011, 17, 343–350.
- Liu, J.; Miao, L.; Sui, J.; Hao, Y.; Huang, G. Nanoparticle Cancer Vaccines: Design Considerations and Recent Advances. Asian J. Pharm. Sci. 2020, 15, 576–590.
- Ai, Y.-Q.; Cai, K.; Hu, J.-H.; Jiang, L.-W.; Gao, Y.-R.; Zhao, H.; Jia, S.-C. The Clinical Effects of Dendritic Cell Vaccines Combined with Cytokine-Induced Killer Cells Intraperitoneal Injected on Patients with Malignant Ascites. Int. J. Clin. Exp. Med. 2014, 7, 4272–4281.
- Geller, M.A.; Knorr, D.A.; Hermanson, D.A.; Pribyl, L.; Bendzick, L.; McCullar, V.; Miller, J.S.; Kaufman, D.S. Intraperitoneal Delivery of Human Natural Killer Cells for Treatment of Ovarian Cancer in a Mouse Xenograft Model. Cytotherapy 2013, 15, 1297–1306.
- Dobrzanski, M.J.; Rewers-Felkins, K.A.; Quinlin, I.S.; Samad, K.A.; Phillips, C.A.; Robinson, W.; Dobrzanski, D.J.; Wright, S.E. Autologous MUC1-Specific Th1 Effector Cell Immunotherapy Induces Differential Levels of Systemic TReg Cell Subpopulations That Result in Increased Ovarian Cancer Patient Survival. Clin. Immunol. 2009, 133, 333–352.
- Deng, J.; Wang, L.; Chen, H.; Li, L.; Ma, Y.; Ni, J.; Li, Y. The Role of Tumour-Associated MUC1 in Epithelial Ovarian Cancer Metastasis and Progression. Cancer Metastasis Rev. 2013, 32, 535–551.
- Alkayyal, A.A.; Tai, L.-H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Makrigiannis, A.P.; et al. NK-Cell Recruitment Is Necessary for Eradication of Peritoneal Carcinomatosis with an IL12-Expressing Maraba Virus Cellular Vaccine. Cancer Immunol. Res. 2017, 5, 211–221.
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028.
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152.
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398.
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108, djv439.
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-Engineered T Cells for Cancer Therapy. Nat. Rev. Cancer 2013, 13, 525–541.
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-Specific T-Cell Activation Independently of the MHC: Chimeric Antigen Receptor-Redirected T Cells. Front. Immunol. 2013, 4, 371.
- Katz, S.C.; Point, G.R.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, R.P. Regional CAR-T Cell Infusions for Peritoneal Carcinomatosis Are Superior to Systemic Delivery. Cancer Gene Ther. 2016, 23, 142–148.
- Ang, W.X.; Li, Z.; Chi, Z.; Du, S.-H.; Chen, C.; Tay, J.C.K.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S. Intraperitoneal Immunotherapy with T Cells Stably and Transiently Expressing Anti-EpCAM CAR in Xenograft Models of Peritoneal Carcinomatosis. Oncotarget 2017, 8, 13545–13559.
- García-Olmo, D.; Villarejo Campos, P.; Barambio, J.; Gomez-Heras, S.G.; Vega-Clemente, L.; Olmedillas-Lopez, S.; Guadalajara, H.; Garcia-Arranz, M. Intraperitoneal Collagenase as a Novel Therapeutic Approach in an Experimental Model of Colorectal Peritoneal Carcinomatosis. Sci. Rep. 2021, 11, 503.
- Koneru, M.; Purdon, T.; Spriggs, D.; Koneru, S.; Brentjens, R. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors in Vivo. Oncoimmunology 2015, 4, e994446.