Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Kondylia Antoniadi and Version 2 by Dean Liu.
Improvements in the treatment of childhood cancer have considerably enhanced survival rates over the last decades to over 80% as of today. However, this great achievement has been accompanied by the occurrence of several early and long-term treatment-related complications major of which is cardiotoxicity.
- cardiotoxicity
- childhood cancer
- chemotherapeutics agents
- biomarkers
1. Cardiotoxicity
The term cardiotoxicity was first described in 1946 as the damage to the heart caused by local anesthetics, mercurial diuretics, and digitalis. Later, in the 1970s, the term broadened to encompass cardiac complications related to anthracyclines (doxorubicin and daunorubicin), combination therapies such as doxorubicin and radiation, and drugs such as 5-fluorouracil. Presently, there is increased research interest, both basic and clinical, in detecting and managing cardiotoxicity as early as possible.
The definition of cardiotoxicity has great significance for a patient’s management. According to the International Cardio-Oncology Society (IC-OS), the cardiovascular complications of chemotherapy can be separated into the following clinical entities and/or categories: (i) cardiac dysfunction: cardiomyopathy/heart failure (HF), (ii) vascular toxicity, (iii) myocarditis, (iv) arterial hypertension, and (v) arrhythmias and QT prolongation [1][2][7,8].
The most preponderant diagnosis of cardiotoxicity is based on the changes found in the left ventricular (LV) systolic function measured by the left ventricular ejection fraction (LVEF). Different organizations have defined cardiotoxicity in several ways using different threshold changes in the LVEF [2][8]. The need to harmonize all these definitions has been met by the International Cardio-Oncology Society (IC-OS) and is supported by the 2022 ESC Guidelines [1][2][7,8].
Cardiotoxicity can be categorized according to the time of presentation as acute, early onset, or late onset. Cardiotoxicity can be reversible if addressed while in the early stages [3][9]. Acute (<1%) toxicity can occur either after administrating a single dose or after a course of chemotherapeutic agents, as long as the onset of clinical manifestations is within the first two weeks following the end of the administration. If presented within the first year of treatment, it is characterized as early onset (1–18%). Late or chronic onset is manifested years or even decades following the treatment [3][9]. The percentage of late-onset cardiotoxicity varies in the literature mainly due to the different definitions used, the detection methods for cardiotoxicity, the population monitored, and the study design. It seems that over 50% of pediatric cancer survivors showed a subclinical decline in myocardial function and over 16% showed symptoms of clinical HF, especially those who had been exposed to anthracyclines [3][9].
Abnormalities in ventricular repolarization and electrocardiographic QT-interval alterations, supraventricular and ventricular arrhythmias, acute coronary syndromes, and pericarditis and/or myocarditis-like syndromes are hallmarks of acute or early onset cardiotoxicity [3][9]. In contrast, asymptomatic systolic and/or diastolic LVD, which can result in dilated cardiomyopathy, is the most typical indicator of chronic cardiotoxicity [3][4][9,10]. Clinical and sub-clinical cardiovascular damage, coronary artery disease, and cerebrovascular events are other conditions linked to treatment-related complications. Survivors had an almost six-fold higher risk of heart failure, a five-fold higher risk of myocardial infarction, a six-fold higher risk of pericardial disease, and an almost five-fold higher risk of valvular abnormalities compared to their siblings [5][6][7][11,12,13].
2. Chemotherapeutic Drugs
2.1. Anthracyclines
Anthracyclines, primarily doxorubicin but also daunomycin, epirubicin, and idarubicin, are some of the most commonly used agents for both hematologic and solid tumors. The basic structure of anthracyclines is that of a tetracyclic molecule with an anthraquinone backbone connected to a sugar moiety by a glycosidic linkage (Figure 1).
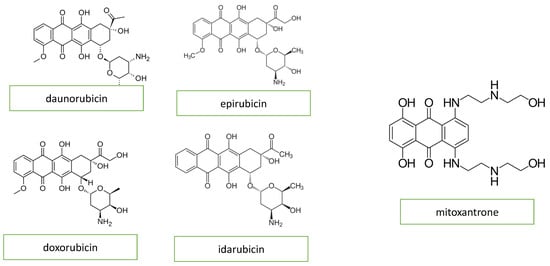
Figure 1. Chemical structure of anthracyclines and mitoxantrone.
Acute cardiotoxicity due to anthracyclines may present as hypotension, tachycardia, arrhythmia, transient depression of left ventricular function, myocarditis, pericarditis, or acute coronary syndrome. Late-onset cardiotoxicity caused by a high cumulative dose of anthracyclines mainly includes signs and symptoms of cardiomyopathy and chronic heart failure [3][9].
Mitoxantrone is a an anthracenedione (1,4-dihydroxy-9,10-anthraquinon, Figure 1) or anthracycline analog and has similar anthracycline mechanisms of action. Mitoxantrone might cause a wide variety of heart conditions, such as disturbances of cardiac rhythm, chronic heart failure, and persistent diastolic dysfunction in the absence of an impairment of the left ventricular ejection fraction [4][10].
The prevalent concept of how anthracycline action may cause heart damage involves the production of oxygen radicals, which in turn damage the DNA, proteins, and lipids, leading to cellular dysfunction and myocyte death [8][9][10][14,15,16].
Cardiolipins are abundantly found on the inner mitochondrial cell membrane. By having an increased affinity for anthracyclines, they in turn allow for their increased cell entry. Upon cell entry by passive diffusion, they can reach much higher intracellular concentrations compared to extracellular compartments. Within the cell, they form complexes by binding to iron, thus producing free radicals and reactive oxygen species, which in turn cause cell damage and death. By peroxidizing lipids of the cell membrane, those elements may also damage the cell membrane. As cardiomyocytes contain an abundance of mitochondria, they are more susceptible to anthracycline damage because of the depletion of glutathione peroxidase (an antioxidant) [9][15].
Other mechanisms of cardiotoxicity include alterations to gene expression and nitric oxide synthase activity, which lead to reduced creatine kinase activity and function in the mitochondria, and ultimately, cell death [9][15]. After exposure to anthracyclines, many of these subcellular sequelae continue to develop for weeks, shedding light on the mechanisms of chronic cardiomyopathy [8][14].
Another identified mechanism of doxorubicin-mediated cardiotoxicity is changes to the topoisomerase-II (Top2). Topoisomerase II (TOP2) is a molecule that anthracyclines bind to and inhibit, preventing the growth of tumors. DNA’s phosphate backbone is broken, twisted, and then resealed by topoisomerases, allowing the double helix’s tension to be changed during transcription and replication. Anthracyclines intercalate into DNA, forming complexes with TOP2 that halt the enzyme’s activity and trigger a DNA damage reaction that results in cell death [8][9][11][14,15,17] (Figure 2).
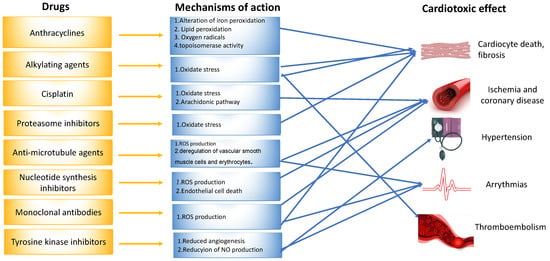
Figure 2. Mechanisms of drug-induced cardiotoxicity.
The mechanisms of mitoxantrone-associated cardiotoxicity remain to be completely understood. The formation of reactive oxygen species in myocardial cells is thought to lead to tissue damage through interactions with cellular iron metabolism [4][10].
2.2. Nucleotide Synthesis Inhibitors
The clinical presentation of methotrexate and fluorouracil (5-FU)-induced cardiotoxicity includes myocardial ischemia, cardiogenic shock, heart failure, and cardiomyopathy [4][10]. Coronary spasm is the most frequently reported mechanism of 5-FU-induced cardiotoxicity (Figure 2) [4][10]. The data derived from animal models indicate that these chemotherapeutic agents induce oxidative stress and the subsequent apoptosis of cardiomyocytes and endothelial cells [4][11][10,17].
2.3. Alkylating Agents
Adjuvant DNA-alkylating agents, such as cyclophosphamide (CP) and ifosfamide (IFO), suspend DNA synthesis in cancer cells. These two agents are similar in structure (Figure 3) and engender a similar pattern of cardiotoxic effects, causing acute heart failure, hemorrhagic myopericarditis, and arrhythmia [4][12][10,18]. CP- and IFO-induced acute cardiotoxicity is attributed mainly to a rise in free oxygen radicals and a lower antioxidant defense mechanism in the myocardium (Figure 2). A recent study by Sayed-Ahmed MM et al. demonstrated that CP- and IFO-induced cardiotoxicity is due to the inhibition of long-chain fatty acid oxidation via the repression of carnitine palmitoyl transferase I and fatty acid binding protein [13][19].
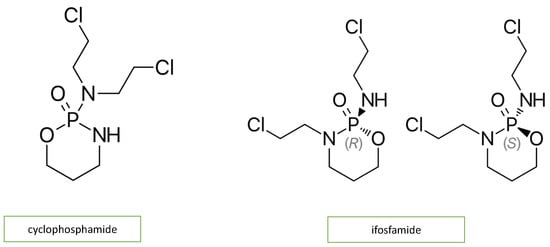
Figure 3. Chemical structure of alkylating agents.
2.4. Tyrosine Kinase Inhibitors
Dasatinib, imatinib, lapatinib, sorafenib, nilotinib, and sunitinib are examples of small molecule tyrosine kinase inhibitors (TKIs) that suppress cancer cell proliferation and induce apoptosis of cancer cells. Imatinib, dasatinib, and nilotinib are the three FDA-approved TKIs for use as first-line chronic myeloid leukemia therapy in pediatrics. In addition, sorafenib is used in young adults. The pathophysiological mechanism of ΤΚΙ-induced cardiotoxicity is mitochondrial impairment and cardiomyocyte apoptosis (Figure 2) [4][12][14][10,18,20]. Each of the above drugs is associated with a different type of cardiotoxicity. For example, dasatinib is more often associated with pleural effusion and less with hypertension, HF, pericardial effusion, and pulmonary hypertension. Nilotinib is associated with peripheral artery disease, hypertension, and prolonged QTc. In contrast, imatinib is related to less cardiotoxicity than the other TKIs [1][7].
2.5. Anti-Microtubule Agents
Anti-microtubule agents, including docetaxel, paclitaxel, and vinca alkaloids, prevent the polymerization or depolymerization of microtubules [11][17]. The clinical features of cardiotoxicity induced by anti-microtubule agents are mostly ischemia and arrhythmia. Among all anti-microtubule agents in clinical use, paclitaxel induces the release of histamine, which in turn activates specific cardiac receptors, raising the myocardium’s oxygen need, and leading to coronary vasoconstriction (Figure 2). In addition, Zhang et al. reported that the frequency of spontaneous calcium concentration in cardiomyocytes was significantly increased after paclitaxel treatment. This finding could be of great significance, as fluctuations in blood calcium levels are linked to arrhythmogenesis [4][15][10,21].
2.6. Cisplatin
Cisplatin is an efficacious chemotherapeutic drug with a strong antitumor effect against a wide range of neoplasms (Figure 4). However, the drug’s acute and cumulative cardiotoxicity, including electrocardiograph (ECG) abnormalities, angina and acute myocardial infarction, hypertension and hypotension, arrhythmias, myocarditis, cardiomyopathy, and congestive heart failure, is a significant factor that restricts cisplatin treatment. Cisplatin cardiotoxicity can be caused by reactive oxygen species generation, which leads to the creation of oxidative stress and endothelial capillary damage (vascular damage) or has a direct toxic effect on cardiac myocytes (Figure 2) [4][10]. The prolonged cardiovascular toxicity of cisplatin, lasting up to many years, has been explained by both direct diffuse endothelial damage and an increase in risk factors for cardiovascular disease. These effects include coronary artery disease, systolic or diastolic left ventricular dysfunction, and severe congestive cardiomyopathy [4][10].
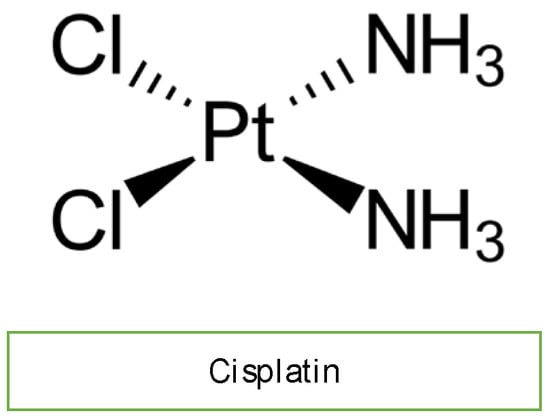
Figure 4. Chemical structure of cisplatin.
2.7. Monoclonal Antibodies
Monoclonal antibodies, including bevacizumab (Avastin) and trastuzumab, not used in children, inhibit angiogenesis. Bevacizumab blocks vascular endothelial growth factor (VEGF), while trastuzumab inhibits human epidermal growth factor receptor 2 (HER2) in cancer cells (Figure 5). Bevacizumab causes mostly hypertension, congestive heart failure, and thromboembolic events of the artery and vein through the mechanism of oxidate stress induced by cardiomyocyte apoptosis. Monoclonal antibodies are not widely used in children with malignancy, so rwesearchers do not discuss them further [4][12][14][10,18,20].
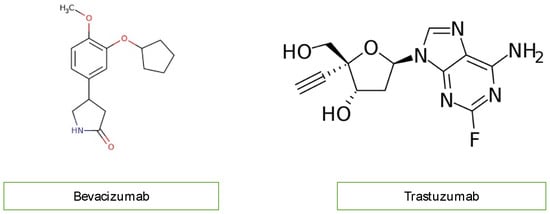
Figure 5. Chemical structure of monoclonal antibodies.
2.8. Proteasome Inhibitors
A new therapeutic option for the treatment of acute lymphoblastic leukemia (ALL) includes proteasome inhibitors (Figure 6) [12][14][18,20]. Bortezomib and carfilzomib are two newly prescribed drugs with the potential to cause cardiac dysfunction [16][22]. Compared to carfilzomib (up to 25%), bortezomib has a lower incidence of heart failure (up to 4%). The pathogenesis of proteasome inhibitor cardiotoxicity is not currently well understood. Exposure to proteasome inhibitors in a prenatal mouse model has shown that they can induce oxidative stress, leading to myocardial dysfunction. Carfilzomib is also known to induce renal toxicity and microangiopathy as a consequence of endothelial dysfunction. Combining these studies reveals a complicated mechanism of cardiotoxicity linked to proteasome inhibitors, including alterations to the heart’s muscle and vasculature, which may be more severe with carfilzomib than bortezomib due to the irreversible nature of the proteasome inhibition of carfilzomib [17][18][23,24].
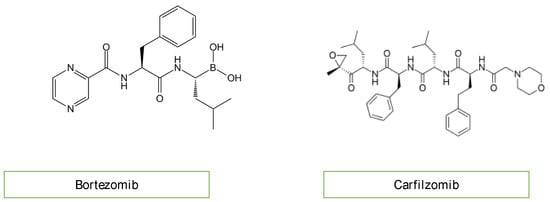
Figure 6.
Chemical structure of proteasome inhibitors.