Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by valerie Vouret-Craviari and Version 2 by Catherine Yang.
P2RX7 belongs to the family of P2X receptors that are assembled and active when in their trimeric form. Each monomer is composed of two transmembrane domains that are connected by a large extracellular loop, and an N- and C- termini domain located intracellularly. However, unlike other members, P2RX7 has a long intracellular C-terminal domain that structurally distinguishes it from the others and confers its unique biological activities. Even though all seven members of the P2X receptors recognize eATP, they are activated with various affinities that range from 0.5 µM for P2RX3 to over 100 µM for P2RX7. Thus, activation of P2RX7 requires high levels of eATP, levels that are found in the tumor microenvironment (TME) which controls the three main activities of the receptor: cationic exchange, macropore opening and NLRP3 inflammasome activation.
- P2RX7
- IL-18
- cancer
1. Cationic Exchange
P2RX7, as well as the other P2X receptors, are non-selective cationic channels that lead to membrane depolarization when bound to eATP. Indeed, activation of P2RX7 leads to calcium and sodium influx as well as potassium efflux, which ultimately activate various signaling pathways that range from cell proliferation to immune-related events [1][11].
It has been reported that low levels of eATP can induce cell proliferation in a P2RX7-dependent manner through calcium influx [2][8]. Indeed, intracellular calcium is a second messenger that controls various cellular functions. It has been shown that the ATP/P2RX7 axis leads to the activation of MAP kinases (ERK1/2, p38, JNK), B kinases (PKB, Akt) and PKC involved in cell proliferation in several types of cancer, in mouse and human models [1][2][8,11]. Moreover, increase of intracellular calcium levels following P2RX7 activation has been shown to promote cancer cell survival via mitochondrial ion homeostasis [3][12], calcineurin activation and NFAT translocation into the nucleus [4][13].
P2RX7-dependent calcium influx has also been shown to be implicated in the immune response. P2RX7 stimulation in mouse microglial cells induced NFAT and NF-κB translocation into the nucleus in a calcineurin-dependent manner, and both are required for the expression of inflammatory cytokines [5][14] and chemokines such as CCL3 [6][15]. On the other hand, the increase of intracellular calcium and extracellular potassium concentrations have been shown to limit B cell activation by decreasing NFAT translocation into the nucleus [7][16]. P2RX7 has also been shown to have a role in immune cell proliferation [8][17]. Indeed, the activation of the ATP/P2RX7 axis in human T cells induces NFAT translocation and IL-2 secretion, necessary for T cell survival and proliferation [9][18]. Accordingly, it has been shown that the expression of P2RX7 on mouse CD8+ T cells enhances mitochondrial functions through calcium influx and AMPK activation, required to support the generation and survival of memory T cells [10][19] and efficiently eradicate tumor cells [11][20]. In line with these findings, the same authors have shown, in viral infections mouse models, that P2RX7 induces the expression of the TGBRII receptor through calcineurin activation, thereby sustaining the generation of tissue-resident memory T cells [12][21] that require TGFβ sensing for their survival. However, a contradictory study had previously shown, in a mouse model of melanoma, that the expression of P2RX7 on tumor-infiltrating lymphocytes has a detrimental role by limiting T cell proliferation and inducing T cell senescence through mitochondrial reactive oxygen species (ROS) and p38 MAPK activation [13][22]. The discrepancy between the studies could be due to a different activation protocol of T cells, as explained by the authors [11][20].
2. Macropore Opening
One particular characteristic of P2RX7 is the capacity to induce cell permeabilization and death, which paved the way to the discovery of the receptor. The receptor was initially named P2Z since high levels of ATP were shown to induce cell death in human macrophages [14][23]. Its cloning in 1996 highlighted that its structure is close to the P2X receptors [15][24], which led to it becoming the seventh member of the P2X family.
The P2RX7-dependent cell permeabilization is linked to its ability to cause the opening of macropores of 8.5 Å on the plasma membrane, leading to cell death [16][25]. The pores allow the passing of macromolecules up to 900 Da in a non-selective and bidirectional manner, disturbing the intracellular homeostasis [16][25]. The nature of this pore is still under debate. Either (1) P2RX7 recruits pore-forming proteins following its activation, such as pannexine-1 or connexine-43 [17][18][19][26,27,28], or (2) the pores are shaped due to the dilatation of the canal formed by the three monomers of P2RX7 [20][21][22][23][29,30,31,32]. Nevertheless, it is certain that the pore-forming activity of P2RX7 requires the presence of 177 aminos acids in the C-terminal domain [15][24], which explains the selective activity of P2RX7, as compared to the other P2X receptors.
Given its ability to induce cell death, one can speculate that the expression of P2RX7 on tumor cells is beneficial. Indeed, studies using tumor cell lines and preclinical mouse models in melanoma [24][25][33,34], non-melanoma skin cancers [26][35], intestinal carcinomas [27][36] and breast cancer [28][37] have shown that activation of P2RX7 reduced cell proliferation by inducing cell death. On the other hand, many reports show an increase of P2RX7 expression in various tumor cell lines and in patients [29][30][31][32][33][38,39,40,41,42] and this increase is linked to tumor growth, as discussed in the previous section. When the human P2RX7 gene is subjected to alternative splicing, a C-terminal-deleted variant named P2RX7-B [34][43] is generated, preventing the receptor from forming macropores. This accords with reports that P2RX7-B is overexpressed in lung adenocarcinomas [35][44], acute myeloid leukemia [36][45], osteosarcoma [37][46], neuroblastoma [38][47] and glioblastoma [39][48]. In addition, a non-functional form of P2RX7 that lacks macropore activity has been reported in several types of cancers including lung and glioblastomas [40][41][49,50]. Whether the expression of the non-functional form is linked to P2RX7-B is currently unknown. However, the existence of isoforms unable to form macropores can explain how a cell death-inducing receptor can favor tumor growth.
Not only is the macropore activity associated with tumor cell death, but also with immune functions. P2RX7 is implicated in the regulation of T cell homeostasis through induction of cell death, which is linked to the levels of expression of the receptor. Indeed, the expression levels of P2RX7 are regulated according to the T cell subset, its activation status as well as its localization as extensively reviewed in [42][43][51,52]. For instance, it has been shown that the expression of P2RX7 is downregulated following the activation of T cells, protecting them from cell death [44][53]. This is the case for follicular T cells in Peyer’s Patches [45][54] and tissue-resident memory T cells [46][47][55,56]. The purpose of P2RX7’s downregulation is not only to prevent T cell death by protecting antigen-reactive T cells, but also to favor the ionic activity of the receptor and to confer a better fitness of CD8+ T cells [10][19], which consequently enhances the control of tumor growth [11][20].
On the other hand, T regulatory cells (Tregs) and natural killer T cells (NKT) express high levels of P2RX7 and are more susceptible to P2RX7-induced cell death [42][48][51,57]. ThWe researchers hhave shown that p2rx7-/- mice with colitis-associated cancer develop bigger and more aggressive lesions than WT mice, which was associated with an accumulation of Tregs within the lesions [49][58]. Moreover, early depletion of Tregs in vivo by activation of P2RX7-expressing T cells using the NAD+/ART2 axis (known to activate P2RX7 in mouse T cells [44][53]) in tumor mouse models increased the anti-tumor effector functions of CD8+ T cells [50][59].
3. NLRP3 Inflammasome Activation
Another feature of P2RX7 that differentiates it from other P2X receptors is its ability to activate the NLRP3 inflammasome. The NLRP3 (NOD-like receptor family, pyrin domain containing 3) protein belongs to the NOD-Like receptors (NLR) family which are pattern recognition receptors (PRR), meaning that NLRP3 is implicated in danger signal recognition [51][60], highlighting its importance in the establishment of the immune response, notably in the context of cancer. It is, therefore, mainly studied in the innate immune cells from the myeloid lineage such as macrophages, monocytes and dendritic cells. This is supported by the high expression levels of P2RX7 on these cells compared to other immune cells [1][52][11,61].
Even though NLRP3 is not a receptor per se, it is a cytosolic sensor of stress that interacts with other proteins to form a multimeric complex of proteins called the NLRP3 inflammasome. The complex involves the NLRP3 protein (containing a pyrin domain), the apoptosis-associated speck-like protein containing a CARD (ASC, with a CARD and pyrin domain) and the effector protein pro-caspase-1 (with a CARD domain). The proteins associate by way of ASC, which acts as an anchor through the respective binding of the CARD and pyrin domains of NLRP3 and pro-caspase-1. The NEK7 protein (NIMA-related kinase 7) is necessary for the oligomerization of the NLRP3 subunits to form the NLRP3 inflammasome [53][62]. The oligomerized NLRP3 inflammasome causes the self-cleavage of the pro-caspase-1 into active caspase-1, which, in turn, cleaves the inactive precursors of the IL-1β and IL-18 inflammatory cytokines intracellularly to shape the immune response.
The ATP/P2RX7 axis is not the only activator of the NLRP3 inflammasome but is described as the most potent [54][63]. Indeed, the NLRP3 inflammasome can also be activated by a variety of stress signals such as pore-forming toxins, ionophores or uric acid crystals [55][64]. It has been reported that the main event leading to the assembly of the NLRP3 inflammasome is potassium efflux, which is common for all its activators, including ATP [55][56][64,65]. Potassium efflux is a common feature of all the P2X receptors as well as other potassium channels, yet they are unable to independently activate the NLRP3 inflammasome. Indeed, the C-terminal domain of P2RX7 is necessary for the activation of NLRP3 by ATP, since its absence prevents NLRP3 activation and IL-1β release [23][32]. Moreover, unlike the other P2X receptors, P2RX7 does not desensitize [57][9], leading to continuous potassium efflux that may facilitate NLRP3 activation. On the other hand, NLRP3 and P2RX7 have been shown to interact at the plasma membrane either directly [58][66] or indirectly via the Paxillin protein [59][67] to induce the assembly of NLRP3. It has also been shown that P2RX7-mediated NLRP3 activation requires the potassic channel TWIK2, where both receptors act in synergy to decrease intracellular potassium levels and activate NLRP3 [60][68]. Besides potassium efflux, P2RX7-mediated calcium influx has also been shown to assemble NLRP3, through ROS production and mitochondrial depolarization [61][69]. Therefore, the potency of P2RX7 to activate NLRP3 could be explained by the fact that P2RX7 can induce calcium influx, potassium efflux and interact with other potassic proteins as well as with NLRP3 itself, leading to a robust activation of the NLRP3 inflammasome.
The role of NLRP3 in cancer progression is not clear. Indeed, there are several conflicting studies in various types of cancer showing that NLRP3 can either favor tumor growth or act as a tumor suppressor [62][63][70,71]. Since the readout activity of NLRP3 is the release of mature inflammatory cytokines (Figure 1), the IL-1β and IL-18 have an important role in cancer progression.
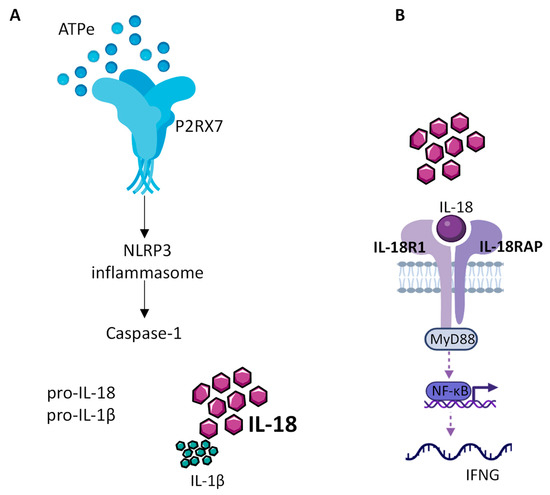
Figure 1. IL-18 production by the NLRP3 inflammasome. (A) Extracellular ATP (eATP), present at high concentration within the TME, activates P2RX7, which in turn allows the assembly and activation of the NLRP3 inflammasome leading to caspase1 activation, cleavage of the constitutive pro IL-18 cytokine and release of mature IL-18. (B) Released mature IL-18 binds to IL-18 receptor, composed of IL-18R1 and IL-18RAP, to activate NF-kB, which in turn controls the production of INF-γ.