Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by 朋恵 藤田 and Version 2 by Lindsay Dong.
The importance of uric acid, the final metabolite of purines excreted by the kidneys and intestines, was not previously recognized, except for its role in forming crystals in the joints and causing gout. Uric acid is not a biologically inactive substance and may exert a wide range of effects, including antioxidant, neurostimulatory, proinflammatory, and innate immune activities.
- dysuricemia
- hyperuricemia
- hypouricemia
1. Introduction
Uric acid was first identified in urine by Scheele in 1776 and later extracted from the tophi of gout by Wollastone in 1787 [1][2]. In 1848, Garrod discovered that patients with gout had elevated blood glucose levels [2][3]. Gout is a type of inflammatory arthritis that occurs when sodium uric acid crystallizes in the joints and is preceded by hyperuricemia. Historically, hyperuricemia was considered synonymous with gout. Uric acid was thought to be a form of waste excreted from the kidneys and intestinal tract. When serum uric acid rises above the dissolution limit, it crystallizes and precipitates in the joints, causing inflammation. If it precipitates in the urinary tract, it forms stones. Therefore, serum uric acid levels were measured only when gouty arthritis and uric acid urolithiasis were suspected. Traditionally, asymptomatic hyperuricemia was generally considered a benign disease that did not require treatment [3][4][4,5]. However, in recent years, it has become increasingly clear that uric acid is not a biologically inert substance but has various biological functions. Therefore, both high and low uric acid levels can affect an organism, even if the condition is asymptomatic.
Hyperuricemia is strongly associated with lifestyle-related diseases such as hypertension [5][6][6,7], type 2 diabetes mellitus [7][8], and metabolic syndrome [8][9]. It may contribute to myocardial infarction, an atherosclerotic disease [9][10][10,11]. In addition, gout caused by hyperuricemia is extremely painful and reduces the quality of life [11][12] and physical function [12][13]. Conversely, a study implied that hypouricemia causes exercise-induced renal failure and urinary stones [13][14]. Therefore, it is now understood that both elevated and reduced uric acid levels can have adverse effects on the living body.
2. Definition and Epidemiology
Hyperuricemia is determined by the solubility of uric acid and is defined as a serum uric acid level greater than 7.0 mg/dL in both men and women [13][14]. In the NHANES, the prevalence rates of gout and hyperuricemia remained substantial in the U.S. between 2007 and 2016, with gout having a prevalence of 3.9% (9.2 million individuals) among U.S. adults from 2015 to 2016. The mean serum uric acid level was 6.0 mg/dL in men and 4.8 mg/dL in women, and the prevalence of hyperuricemia was 20.2% in men and 20.0% in women [14][1]. The data show that the prevalence of hyperuricemia has been stable over the last 10 years, but it still persists at a considerable rate. Although there is no established definition of hypouricemia, a value of 2.0 mg/dL or less is generally used as a reference value for this condition [13][14]. Hypouricemia is less common than hyperuricemia, and in Japan, its prevalence was reported to be 0.2% in men and 0.4% in women [15]. Hypouricemia was also found to be a rare outcome in Germany, affecting 0.09% of the population (0% in men; 0.27% in women) [16]. Regardless of race or region, hypouricemia has been found to be more prevalent in women than in men.2.1. Serum Uric Acid Regulation
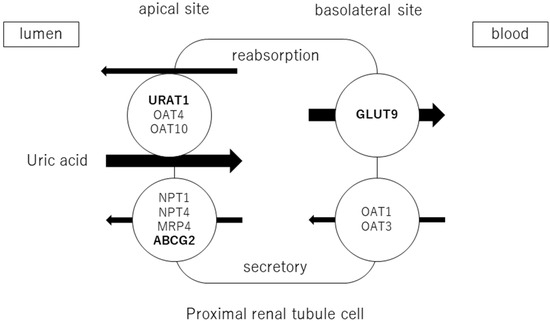
Blood uric acid levels depend on the balance between the exogenous production of uric acid via dietary factors (including purine intake) and its endogenous production via purine metabolism, reabsorption and excretion by the kidney, and excretion by the intestine [17]. Thus, uric acid is homeostasis through the complicated process of production, secretion, and reabsorption in the kidney and excretion in the intestines. Uric acid is produced by the metabolism of endogenous purines (approximately 300–400 mg synthesized daily) and exogenous purines (approximately 300 mg from the diet), totaling 1200 mg in healthy men (600 mg in healthy women) on purine-free diets. The purine nucleobases adenine and guanine are components of both ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). Therefore, the breakdown of RNA and DNA increases blood uric acid levels. Many hematological diseases, such as acute myelogenous leukemia, involve the production of excessive uric acid as tumor cells proliferate and collapse, causing hyperuricemia. Hyperuricemia is also caused by the rapid disintegration of tumor cells by antitumor agents. This is called tumor lysis syndrome and is complicated by hyperkalemia and hyperphosphatemia. Hyperuricemia associated with hematological diseases reflects tumor volume. It may also indicate a response to treatment with antitumor agents. Rapidly elevated uric acid levels can lead to renal failure, which is treated with uric acid inhibitors or uric acid-degrading enzymes, depending on the risk of developing the disease [18]. In addition, uric acid is produced by the breakdown of adenosine triphosphate (ATP) through the metabolism of fructose and alcohol. ATP is an important source of energy for intracellular reactions. In terms of dietary effects, animal protein contributes significantly to the intake of this purine. In addition, serum uric acid levels are increased by the intake of glutamic acid, which is metabolized to uric acid in the liver, and by the intake of foods high in purines and uric acid itself. Foods that affect serum uric acid levels include alcohol, meat and seafood, soft drinks, dairy products, coffee, and vitamin C. A purine-free diet reduces uric acid excretion via urine by approximately 40% [19], indicating that dietary purine contributes greatly to serum uric acid levels. Adenosine products are converted to hypoxanthine, and hypoxanthine is oxidized to xanthine and then uric acid by xanthine oxidoreductase (XOR). Approximately two-thirds of uric acid is excreted by the kidneys and one-third by the small intestine. Uric acid is filtered through the glomeruli and reabsorbed in the proximal tubules. The excretion rate of uric acid in the proximal tubules of the kidney is approximately 10% [20] and is regulated by transporters expressed at the apical and basolateral sides. Uric acid reabsorption is mediated by URAT1/SLC22A12, OAT4/SLC22A11, and OAT10/SLC22A3 transporters at apical sides [21], and by GLUT9/SLC2A9 transporter at basolateral sides. Uric acid secretion is mediated by OAT1/SLC22A11 and OAT3/SLC22A8 transporters at the basolateral sides and by NPT1/SLC17A1, NPT4/SLC17A3, MRP4/ABCC4, and BCRP/ABCG2 at the apical sides [22][23][22,23] (Figure 1). URAT1 and GLUT9 are transporters for uric acid reabsorption, and their dysfunction causes hypouricemia. Patients with URAT1 and GLUT9 dysfunctional variants are called hereditary renal hypouricemia types 1 and 2, respectively. In the intestine, BCRP/ABCG2 secretes uric acid into the intestinal lumen [24]. Genome-wide association studies have reported over 30 genetic variants in uric acid transporters that affect serum uric acid. Three uric acid transporters, URAT1, GLUT9, and ABCG2, play key roles in the regulation of serum uric acid, and their dysfunctions lead to dysuricemia (hypouricemia and hyperuricemia). The role of ABCG2 variants has been shown to be more important for the risk of hyperuricemia than environmental factors such as obesity and intense alcohol consumption [25]. Nonsynonymous allelic variants of ABCG2 were shown to significantly hasten the onset of hyperuricemia and increase the likelihood of gout and the presence of a family history of gout. ABCG2 dysfunction was known to be a risk factor for pediatric-onset hyperuricemia and gout [26].Figure 1. Uric acid transport molecules in the proximal renal tubules. Uric acid is regulated by several transporters expressed at the apical and basolateral sides. Uric acid reabsorption is mediated by URAT1/SLC22A12, OAT4/SLC22A11, and OAT10/SLC22A3 transporters at the apical side, and GLUT9/SLC2A9 transporter at the basolateral side. Uric acid secretion is mediated by OAT1/SLC22A11 and OAT3/SLC22A8 transporters at the basolateral side, and NPT1/SLC17A1, NPT4/SLC17A3, MRP4/ABCC4, and BCRP/ABCG2 at the apical side. URAT1, GLUT9, and ABCG2 were bolded, as they play crucial roles in the regulation of serum uric acid, and their dysfunctions cause uric acid transport disorders (hypouricemia and hyperuricemia).
2.1.1. The Pros and Cons of Uric Acid: The Pros
Over the course of human evolution, mutations in genes involved in ascorbic acid synthesis caused loss of function. Simultaneously, mutations in the uricase gene led to uric acid becoming the final metabolite in this pathway. Ames et al. showed that uric acid might act as an antioxidant in various redox reactions [27][36]. Uric and ascorbic acids are considered the most important water-soluble antioxidants [28][37], and the plasma level of uric acid is approximately six times that of ascorbic acid [29][38]. Ascorbic acid has a tendency to undergo oxidization and mutation, suggesting that the resulting mutated metabolite may contain peroxide radicals. Therefore, uric acid is considered a better antioxidant than ascorbic acid [30][39]. In addition, Yeum et al. measured the antioxidant capacity of the human body and suggested that uric acid accounts for 60% of the total antioxidant capacity in plasma [31][40]. This suggests that uric acid may provide the major endogenous defense against oxidative damage in the body [32][41]. Fabbrini showed that uric acid is a major antioxidant that may protect against oxidative damage caused by free radicals [33][42]. It is widely accepted that high levels of blood uric acid are an evolutionary advantage in humans because uric acid protects the heart, blood vessels, and nerve cells from oxidative damage. Uric acid also acts as a defense against aging and cancer by preventing oxidative damage caused by ROS [34][43] (Figure 2).
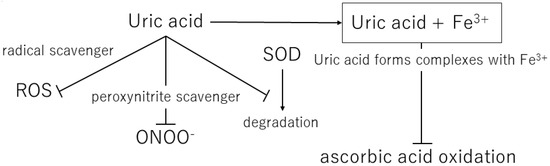
Figure 2. Mechanisms of the antioxidant effects of extracellular uric acid. ROS, reactive oxygen species; ONOO−, peroxynitrite; SOD, superoxide dismutase.
Uric acid reacts with a variety of oxidants, among which it has been reported that it preferentially reacts with peroxynitrite to form triuret [35][44]. Peroxynitrite is thought to contribute to the development of cardiovascular disease and has been shown to cause oxidative damage via tissue nitration [36][45]. Uric acid has been suggested to act as a scavenger of peroxynitrite [37][46]. In addition, uric acid can help keep the levels of superoxide dismutase, an enzyme playing a key role in protecting the extracellular environment from oxidative stress. In the extracellular environment, uric acid can scavenge hydroxyl radicals and peroxynitrite, but it has been shown to lose antioxidant capacity in a hydrophobic environment [38][39][47,48].
2.1.2. The Pros and Cons of Uric Acid: The Cons
Uric acid produces allantoin and peroxynitrite via superoxide radicals to form triuret. The chemical reaction of uric acid with peroxynitrite produces aminocarbonyl and triurethanol radicals, which react with myeloperoxidase to produce the pro-oxidant uric acid hydroperoxide [40][58]. In contrast, uric acid in cells stimulates reduced NADPH oxidase and functions as a pro-oxidant [37][46] (Figure 3). The effects of exogenous uric acid on cells can be prevented by inhibiting the uptake of uric acid into the cells. Similarly, the biological effects of endogenous uric acid can be prevented by inhibiting its synthesis using XOR inhibitors [41][59]. Uric acid exhibits autocrine, paracrine, and endocrine activities. Intracellular uric acid stimulates proinflammatory transcription factors, growth factors, vasoconstrictive substances (angiotensin II, thromboxane, and endothelin), and chemokines, leading to mitochondrial dysfunction [40][41][58,59]. Uric acid also reduces endothelial NO bioavailability through various mechanisms, causing endothelial cell damage [42][43][60,61]. Thus, depending on the chemical environment, uric acid may not be an antioxidant but a pro-oxidant.
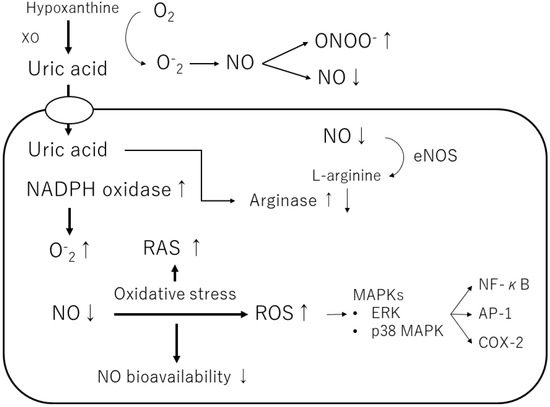
Figure 3. Mechanisms of the pro-oxidant effect of intracellular uric acid. Intracellular uric acid induces reactive oxygen species (ROS) production and activates several signaling pathways. XO, xanthine oxidase; NO, nitric oxide; ONOO−, peroxynitrite; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase; eNOS, endothelial NO synthase; ERK, extracellular signaling-regulated kinase; MAPK, mitogen-activated protein kinase; RAS, renin-angiotensin-aldosterone system; NF-κB, nuclear factor-kappa B; AP-1, activator protein 1; COX-2, cyclooxygenase 2.
Furthermore, uric acid induces the growth of vascular smooth muscle cells (VSMCs) and triggers activation of the extracellular signal-regulated kinase (ERK), mitogen-activated protein (MAP) kinases, and platelet-derived growth factor (PDGF). Uric acid also induces the inflammatory pathway in VSMCs and increases the production of monocyte chemoattractant protein-1 (MCP-1). It has also been shown to cause the activation of cyclooxygenase-2 (COX-2), p38 MAPK, nuclear transcription factor nuclear factor-kappa B (NF-κB), and activator protein-1 (AP-1) [40][58]. According to a previous study, soluble uric acid, as well as uric acid crystals, can cause inflammation.
2.2. Hypertension
Numerous epidemiological studies have reported that elevated uric acid is an independent predictor of hypertension [44][62]. The relationship between serum uric acid levels and blood pressure was found to be dose-dependent and linear. Hyperuricemia is a strong independent predictor of hypertension with an approximately 2-fold increased risk within 5–10 years [45][46][47][63,64,65]. Hyperuricemia induced by uricase inhibitors in rats leads to hypertension. The mechanism by which hyperuricemia causes hypertension involves the sympathetic nervous system hyperactivity, endothelial dysfunction with reduced NO levels, and renal vasoconstriction mediated by activation of the renin-angiotensin system [48][66].
In a study of new-onset essential hypertension by Feig et al., 90% of subjects with essential hypertension and 30% of those with secondary hypertension had elevated serum uric acid levels (>5.5 mg/dL) compared with 0% of controls, revealing a clear association between uric acid and hypertension [49][69]. Kuwabara et al. also found that, for every 1 mg/dL decrease in serum uric acid in healthy male subjects, the protective effect against hypertension increased by 18%. In healthy female subjects, each 1 mg/dL decrease in serum uric acid levels was associated with a 31% decrease in the development of hypertension [50][70].