Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Guodong Zhu and Version 2 by Fanny Huang.
Kidney tumors comprise a broad spectrum of different histopathological entities, with more than 0.4 million newly diagnosed cases each year, mostly in middle-aged and older men. Based on the description of the 2022 World Health Organization (WHO) classification of renal cell carcinoma (RCC), some new categories of tumor types have been added according to their specific molecular typing. However, studies on these types of RCC are still superficial, many types of these RCC currently lack accurate diagnostic standards in the clinic, and treatment protocols are largely consistent with the treatment guidelines for clear cell RCC (ccRCC), which might result in worse treatment outcomes for patients with these types of molecularly defined RCC.
- renal cell carcinoma
- molecular
- treatment
- clinical
1. Introduction
Kidney cancer is the 14th most common cancer worldwide, and its incidence has continued to increase in recent years [1]. To date, more than 0.4 million new cases of kidney cancer are diagnosed each year [2][3][2,3]. Among them, more than 85% of patients present with renal cell carcinoma (RCC) [4]. Based on the traditional histopathological classification, RCC can be divided into three main categories: clear cell carcinoma (ccRCC, 75%), papillary renal cell carcinoma (PRCC, 15–20%), and chromophobe cell renal carcinoma (chRCC, 5%) [5]. Studies in recent years have found that RCC mostly occurs in older men [6] and most cases are localized tumors, with only 17% of RCC patients having distant metastases at the time of diagnosis, which are mainly found in lung, bone, liver, lymph nodes, and adrenal gland [1][4][1,4]. In 2020, a statistic by Padala SA showed that the 5-year survival rate for kidney cancer patients with metastatic disease was only 12% [7]. Currently, there are more and more treatment modalities for patients with metastatic RCC (mRCC), with targeted therapies and immune checkpoint inhibitor-based immunotherapy gradually proving to be effective in the treatment of patients with mRCC and the survival rate of those patients greatly improving recently [8][9][8,9].
Epigenetic alterations are considered to be a hallmark of cancer [10]. However, recent studies found that RCC has multiple molecular alterations, such as DNA methylation and micro-RNA alterations in ccRCC, which could greatly affect the biological progression of these tumors [11][12][11,12]. The 2022 World Health Organization (WHO) classification of pathological kidney tumors added new histopathological subtypes, including molecularly defined RCC [5][8][5,8]. It includes transcription factor binding to IGHM enhancer 3 (TFE3)-rearranged renal cell carcinomas, transcription factor EB (TFEB)-altered renal cell carcinomas, elongin C (ELOC)-mutated renal cell carcinoma, fumarate hydratase (FH)-deficient renal cell carcinoma, succinate dehydrogenase (SDH)-deficient renal cell carcinoma, anaplastic lymphoma kinase (ALK)-rearranged renal cell carcinomas, and SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1)-deficient renal medullary carcinoma (see Table 1) [5]. These different molecularly defined histopathological subtypes of RCC are easily confused and may lead to suboptimal treatment outcomes as a result of misdiagnoses [12].
Table 1. Genes of molecularly defined renal cell carcinoma and associated clinical syndromes.
| Molecularly Defined Renal Cell Carcinoma Types | TFE3-Rearranged Renal Cell Carcinomas | TFEB-Altered Renal Cell Carcinomas | Elongin C (ELOC, Formerly TCEB1)-Mutated Renal Cell Carcinoma | Fumarate Hydratase-Deficient Renal Cell Carcinoma | Succinate Dehydrogenase-Deficient Renal Cell Carcinoma | ALK-Rearranged Renal Cell Carcinomas | SMARCB1-Deficient Renal Medullary Carcinoma |
|---|---|---|---|---|---|---|---|
| Mutated genes | Transcription factor binding to IGHM enhancer 3 (TFE3) | Transcription factor EB (TFEB) | Elongin C (ELOC) | Fumarate hydratase (FH) gene | Succinate dehydrogenase (SDH) | Anaplastic lymphoma kinase (ALK) | Subfamily B member 1 (SMARCB1) |
| Location of genes | Xp11.23 | 6p21 | 8q21.11 | 1q43 | SDHA: 5p15 SDHB: lp35-p36.1 SDHC: 1q21 SDHD: 11q23 |
2p23 | 22q11.2 |
| Prevalence age | Childhood | Childhood | Middle and old age | Adult | All ages | Childhood | Teenage |
| Clinical Syndromes | None | None | None | Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) | SDH-deficient tumor syndrome | None | Rhabdoid tumor predisposition syndrome; familial schwannomatosis syndrome |
| Chaperone genes | ASPL, PRCC, SFPQ, CLTC, PARP14, RBM10, NONO, MED15 | MALAT1, CLTC, KHDRBS2, CADM2 | None | None | None | VCL, TPM3, EML4, STRN, HOOK1 | None |
| Mode of inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance | Dominant inheritance |
| Morphological characteristics | Transparent eosinophils; papillary architecture and psammoma bodies under the microscope | TFEB-translocated RCC: the biphasic growth pattern consisting of large and small tumor cells; smaller cells around the basement membrane-like structures; extensive hyalinization; papillary architecture; clear cell morphology. TFEB-amplified RCC: above pattern was less common |
A clear cellular morphology under the microscope; thick fibromuscular bands; branching glandular vesicular; tubular structures | The papillary type or solid, tubulocystic, sieve-like type; abundant eosinophilic granulocytes, perinuclear halo | Cuboidal tumor cells, nested or tubular growth pattern. Characteristic morphology: the presence of vesicles or flocculent inclusions in the cytoplasm |
ALK-rearranged RCC with VCL as a fusion gene: sickle-cell trait; eosinophilic granulocytic stroma; cytoplasmic lumen. Other ALK-rearranged RCC: similar to PRCC; consist of abundant intracellular and extracellular mucins; eosinophilic granuloplasm |
At a high grade at the time of detection; infiltrative growth; sieve or reticular appearance |
| Ancillary test (IHC, FISH) | Positive: PAX8 (100%); TFE3 (95%); CD10 (89%); achromatase (82%). Negative: cytokeratin 7 (CK7); carbonic anhydrase 9 (CA9); GATA3 |
Positive: histone K; Melan-A TFEB-amplified RCC: diffusely or patchily positive when tested for TFEB levels |
Positive: CK7; ELOC; CA9; CD10; ELOC in the nucleus. | Positive: PAX8; succinate dehydrogenase B abnormal succinate semicarbonate (2SC)S-(2-succino)-cysteine. Negative: FH; CK7; TFE3 |
Positive: PAX8; epithelial membrane antigen (EMA). Negative: SDHB; CK7; CD117; histone K; TFE3; HMB45. SDHA-deficient RCC showed negativity for SDHA |
Positive: PAX7; CK10; AMACR; CD3; cytokeratin; ALK. Negative: carbonic anhydrase IX; TFE45; histone enzyme K; Melan A; HMB45 |
Negative: SMARCB1 |
| Oncological behavior and prognosis | May develop metastases within 20–30 years after diagnosis | TFEB-amplified RCC had higher tumor aggressiveness than TFEB-rearranged tumors. The 5-year survival rate for TFEB-amplified RCC was 48% |
Has an aggressive oncological behavior | Have highly staged or distant metastases when diagnosed | Most cases are low grade and have a good prognosis with a low probability of metastasis | ALK-rearranged RCC with VCL as a fusion gene: no recurrence or distant metastasis. Other ALK-rearranged RCC: more aggressive clinical course |
Often found at an advanced stage or with distant metastases; highly aggressive nature of the tumor. Average overall survival: 6–8 months |
2. TFE3-Rearranged Renal Cell Carcinomas
Transcription factor binding to IGHM enhancer 3 (TFE3) is an important regulator of the immune system and has now been shown to cooperate with transcription factor EB (TFEB) to control and regulate carbohydrate and lipid metabolism and mitochondrial homeostasis [13]. The TFE3/TFEB rearrangement renal cell carcinoma is characterized by translocations involving the TFE3 and TFEB genes. They are both derived from the microphthalmia transcription (MiT) family of heterotopic RCC according to the 2016 version of the WHO classification. The MiT subfamily of transcription factors includes TFE3, TFEB, TFEC, and MITF [14]. TFE3- and TFEB-rearranged RCC accounts for 1–4% of the newly diagnosed adult patients [15]. Recent studies have shown that TFE3/TFEB-rearranged RCC can be frequently detected in children [16]. In adults RCC patients, TFE3 ectopic fusions with chaperone genes are more commonly seen [17], and there are no significant prognostic gender differences [15] (Figure 1). This ectopic fusion with a chaperone gene and the decreased immunity in adults TFE3-rearranged RCC patients cause them to have a potentially more aggressive course compared to the pediatric patients [16]. Current studies suggest that previous exposure to cytotoxic chemotherapy might be a predisposing factor [18].
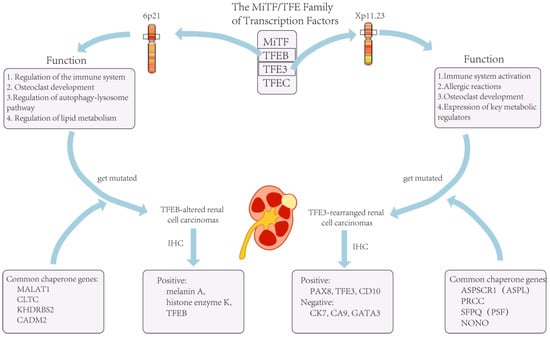
Figure 1. The role of TFE3 in the organism and tumors caused by its mutation.
The list of chaperone genes has been growing and evolving, with more than a dozen having been reported [18]. The three most common translocations currently include a fusion of the PRCC and TFE3 genes, a fusion of the ASPL (ASPSCR1) and TFE3 genes, and a fusion of the SFPQ and TFE3 genes [14]. In addition to this, there are also genes such as CLTC, PARP14, RBM10, NONO, and MED15 that can be fused with ectopic TFE3 [19]. However, current studies suggest that different chaperone genes may exhibit different oncological behaviors and tumor morphologies, and these features vary depending on the type of the involved chaperone genes [18]. For example, TFE3 is more likely to exhibit lymph node metastasis when fused with PRCC than when fused with ASPSCR3 [20].
In terms of histopathological morphology, the characteristics of TFE3 fusion usually presents with transparent eosinophils, a papillary architecture, and psammoma bodies under the microscope [17][21][17,21]. However, due to chaperone genes, RCC with TFE3 rearrangement may also resemble other types of RCC, including ccRCC, PRCC, and epithelioid vascular smooth muscle lipoma [14]. Therefore, attention should be paid and the impact of the genes that are fused with should be determined as much as possible both in the diagnosis and in the treatment of TFE3-rearranged renal cell carcinoma.
When facing TFE3-rearranged renal cell carcinoma, immunohistochemistry (IHC) is the most commonly used examination for diagnosis [13]. If IHC is not used at the time of diagnosis, a large proportion of TFE3-rearranged renal cell carcinomas are likely to be misdiagnosed as ccRCC [19]. For most other types of RCC, the positive IHC markers are cytokeratin 7 (CK7), carbonic anhydrase 9 (CA9), and GATA3. However, these are not expressed in TFE3-rearranged RCC and are usually positive for histone K [19]. In a recent review article, IHC data from nearly 400 cases of TFE3-rearranged RCC patients were analyzed, and the biomarkers with the highest probability of positivity were found to be PAX8 (100%), TFE3 (95%), CD10 (89%), and achromatase (82%) [22]. However, TFE3-rearranged RCC did not always exhibit TFE3 overexpression, and lower TFE3 expression at the time of detection often resulted in false-positive or false-negative results; thus, this could limit the sensitivity and specificity of IHC for detecting TFE3-rearranged RCC [23]. In addition to this, the accuracy of IHC might be affected by the technique and be influenced by the formalin fixation time [18]. On the other hand, there has been no consensus or standardized guidelines regarding the judgment of TFE3 staining results [18], and different pathologists might give completely opposite judgments if specimens show heterogeneous or focal staining. Lee HJ et al. used tissue specimens from 303 RCC patients for IHC testing and found that 23.2% of IHC-negative TFE3 tumors were eventually diagnosed as TFE3-rearranged RCC [24]. Therefore, in clinical practice, a negative TFE3 IHC result alone did not exclude the possibility of a TFE3-rearranged RCC case. Thus, in some cases, a combination of clinical presentation and other examination results might be needed.
The current literature suggests that the detection of TFE3 gene rearrangements by fluorescence in situ hybridization (FISH) is more sensitive and advantageous in experimental manipulation than traditional IHC [23], and its results are more stable in formalin-fixed tissues [14]. Therefore, FISH is currently considered as the gold standard for the diagnosis of TFE3-rearranged RCC [23]. However, some chaperone genes, such as NONO, RBM10, and GRIPAP1, after fusing with TFE3, may not be detected by traditional FISH assays for significant TFE3-positive results. [18]. In addition, similar to IHC testing, the current standard definition of a positive FISH result varies widely among laboratories, from as low as 10% up to 30% [17]. These results suggest that, although the FISH test is currently the gold standard for the diagnosis of TFE3-rearranged RCC, in clinical practice, it should be carefully used together with other test results. For example, the previously mentioned IHC and FISH tests should be considered along with the option of gene probes or alternative molecular techniques [18]. In fact, FISH cannot provide information about fused genes, so in order to further confirm the diagnosis in clinical practice, RNA sequencing is often used to identify the gene involved in the translocation [17]. Recently, TRIM63 determination by RNA in situ hybridization (RNA-ISH) was proposed as an alternative diagnostic tool for TFE3- and TFEB-rearranged RCC [25], but no strong evidence is available from in vitro studies.
Due to the rarity of TFE3-rearranged RCC and the fact that it has not been previously considered as a specific tumor subtype, there are no treatment recommendations for it to date [18]. Most previous treatment regimens are consistent with those for patients with ccRCC; however, due to recent developments in detection technology, its diagnosis has become more accurate, similarly to the detection of ccRCC. More importantly, drugs that normally treat ccRCC may not be effective against TFE3-rearranged RCC [16]. Additionally, Aldera AP et al. found that patients with TFE3-rearranged RCC may develop metastases within 20–30 years after diagnosis, so such patients may also need long-term clinical follow-up [26].
3. TFEB-Altered Renal Cell Carcinomas
As previously stated, TFEB-altered RCC has been included in the MiTF-translocated carcinoma family and TFEB-overexpressing renal tumors were initially identified in pediatric patients. Nowadays, with the availability of accurate examinations, more and more adult RCC patients are diagnosed with TFEB-altered RCC [27]. Nevertheless, the number of TFEB-altered RCC cases is still much lower than for TFE3-rearranged RCC [5]. There are two types of TFEB-altered RCC, including TFEB-rearranged RCC and TFEB-amplified RCC. The TFEB gene in TFEB-rearranged RCC is located on chromosome 6 and is most often translocated into chromosome 11, fusing with the MALAT1 gene. Therefore, it was previously called t(6;11) RCC [19]. In the last few years, researchers have identified cases of RCC related to TFEB amplification, and after further testing and analysis, it was found that both genetic alteration patterns could co-exist in one case [5]. Due to the rarity of the disease, there are few studies on the distinction between different subtypes of TFEB-altered RCC, and current case studies show that the mean age of diagnosis for TFEB-amplified RCC is 62.5–64 years, while the mean age of diagnosis for TFEB translocated RCC is 32.8–34 years [28][29][28,29].
Similar to TFE3-rearranged RCC, TFEB can also be ectopically fused to chaperone genes [28] (Figure 1). Furthermore, for TFEB-amplified RCC, in addition to the possible elevated expression of TFEB, they are often accompanied by the amplification of other oncogenes, such as vascular endothelial growth factor A (VEGFA) and G1 S specific cyclin D3 (CCND3) [30]. It has been shown that these two genes are associated with aggressive oncological behavior [27], which would precisely explain the severe clinical symptoms and poor prognosis of patients with TFEB-amplified RCC.
Although TFEB genes are altered in TFEB-altered RCC, the characteristics of tumor growth vary considerably between different patterns of alteration. It has been widely reported that in TFEB-translocated RCC, the most commonly found morphology is a biphasic growth pattern consisting of large and small tumor cells [27], with smaller cells around the basement membrane-like structures. In addition to this, extensive hyalinization, a papillary architecture, and a clear cell morphology can be seen [31]. However, in TFEB-amplified RCC, this pattern is less common. Gupta S et al. investigated 37 patients with TFEB-altered RCC and found that nearly half of the patients had renal tubular structures and prominent cytoplasmic eosinophilia of tumor cells in their tumor specimens [27].
IHC and FISH are commonly used tests to detect TFEB-altered RCC; however, when assessing whether the TFEB gene is amplified or translocated, the markers used in the detection are quite similar. For TFEB-altered RCC, it has been shown that the staining results for both histone K and Melan-A are positive [31]. Similarly, Gupta S et al. and Wyvekens N et al. studied TFEB-amplified RCC and TFEB-translocated RCC, respectively, and they found that both types of tumors typically express melanin A and histone enzyme K. The difference was that tumor cells in TFEB-amplified RCC were usually diffusely or patchily positive when tested for TFEB levels [27]. However, there was also a subset of TFEB-amplified RCC that had lower TFEB expression levels than TFEB-translocated RCC [29]. Therefore, the type of TFEB gene alteration cannot be distinguished by a TFEB-specific assay alone. If a type of TFEB gene alteration is suspected, it should also be demonstrated using a FISH breakdown test or identified by RNA sequencing with a gene fusion examination [31]. In clinical practice, such detailed testing and diagnosis is not always necessary for all patients because of the very low incidence of the disease, the high cost of FISH, and the use of sequencing tests.
In addition to this, it has also been found that TFEB-amplified RCC exhibits a higher tumor aggressiveness than TFEB-rearranged tumors, and the 5-year survival rate for TFEB-amplified RCC is only 48% [32], while TFEB-translocated RCC progresses more slowly than TFE3-rearranged RCC. Therefore, in clinical practice, physicians should distinguish TFEB-amplified RCC from TFEB-translocated RCC. Since TFEB-altered RCC has often been previously diagnosed as ccRCC, its current treatment modality still differs little from the standard treatment for patients with ccRCC, which may also contribute to the poor prognostic outcome for patients with TFEB-altered RCC.