Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fatima Tuz Zahra and Version 2 by Rita Xu.
The tropylium ion is a non-benzenoid aromatic species that works as a catalyst. This chemical entity brings about a large number of organic transformations, such as hydroboration reactions, ring contraction, the trapping of enolates, oxidative functionalization, metathesis, insertion, acetalization, and trans-acetalization reactions. The tropylium ion also functions as a coupling reagent in synthetic reactions. This cation’s versatility can be seen in its role in the synthesis of macrocyclic compounds and cage structures.
- tropylium cation
- tropylium tetrafluoroborate
- organo-catalyst
1. Introduction
The tropylium ion is a cyclic, planar organic cation with the chemical formula C7H7+. It is formed by the protonation of tropine, which is a bicyclic organic compound found in plants such as Atropa belladonna and Hyoscyamus niger.
The tropylium ion has six π-electrons in its conjugated ring system, which gives it aromatic properties. The structure of the ion is reminiscent of the cycloheptatrienyl anion (tropylium’s isoelectronic counterpart) and is stabilized by the delocalization of the positive charge over the seven carbon atoms of the ring.
The tropylium ion is an important intermediate in the synthesis of many alkaloids, such as atropine and cocaine. It is also used as a spectroscopic probe in organic chemistry research.
The generation of stable and neutral aromatic systems [1] such as benzenoid frameworks [2] has been the driving force [3] for innumerable organic reactions [4] in synthetic chemistry [5]. Their lesser-known analogs, the non-benzenoid [6] carbocyclic aromatic structures [7], used to be considered far less synthetically valuable. These fundamental chemical species [8] usually possess a net charge [9] and are likely to be less stable than benzenoid systems [10].
Merling’s discovery of the bromide salt of the cycloheptatrienyl cation [11], also known as the tropylium ion (1b), in 1891 led to the creation of non-benzenoid carbocyclic aromatic ions [12]. The tropylium ion (1b) is a seven-membered aromatic ring [13] (C7H7+) utilized in chemical synthesis [14] and as a metal complex ligand [15]. The non-benzenoid aromatic tropylium ion [16] (1b) has a 6π-electron system and possesses a positive charge entirely delocalized on a conjugated planar seven-carbon system [17]. As a result, it satisfies Hückel’s aromaticity [18] rule and has a unique combination of stability and reactivity [19].
Nguyen’s research group has made significant contributions to tropylium ion chemistry [20]. He reasoned that the carbocation with one positive charge delocalized [21] over the conjugated seven-membered ring potentially serves as a “soft Lewis catalyst” [22]. Salts of this ion are known to be relatively stable in the solid state or in solutions [23]. It is used in reactions in the form of various salts, such as tropylium tetrafluoroborate (1c) and tropylium perchlorate (1d) (Scheme 1).
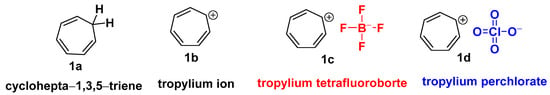
Scheme 1. Structural representation of tropylium ion and its salts (1c) and (1b).
This aromatic ion can be reversibly converted to its non-aromatic neutral structure form by associating with the counterion. As the generation of stable aromatic systems can provide the driving force for many types of chemical transformations, it was believed that the equilibrium between the tropylium ion (1b) and its neutral form could play a vital role in the promotion of organic reactions [24]. Recently, the idea of utilizing this system to facilitate chemical reactions has evolved into versatile methodologies. To date, there have been numerous reports on the applications of non-benzenoid aromatic tropylium ions to accelerate chemical reactions [25].
Due to the reactivity of charged systems, (1b) is capable of forming a covalent bond with other substrates to produce reactive intermediates for further chemical transformations [26]. The reactive intermediates tend to re-aromatize, hence providing the driving force for chemical reactions [27]. The unique combination of stability (due to aromaticity) and reactivity (due to the associated net charge) equips tropylium ions with the unique capability to promote chemical processes [28]. The mechanism of activation by the tropylium ion is potentially applicable to an extended range of substrates and reactions [29]. Furthermore, it can also be used as an organocatalyst, as its neutral de-aromatized counterparts can often be easily converted back to the original aromatic form in situ by the addition of activating reagents [30]. Recently, metal catalysts have been replaced by organocatalysts [31] owing to better tolerance to moisture and air, as well as excellent compatibility with a diverse variety of substrate functional groups [32] and the contribution to green chemistry. This resviearchw focuses on the recent roles of the tropylium (positively charged, 7C, 4n + 2 = 6π-electrons) ion in synthetic chemistry [33].
Tropylium ions (1b) and their salts can be synthesized by a variety of different methods.
Under optimal conditions, the reaction of 4-substituted-4-pentenal derivatives (2a) with (2b) synthesized from pyruvic ester produced 1,6-di- and 1,6,7-trisubstituted-1,3,5-cycloheptatrienes (A) in reasonable yields. The synthesis is attractive since it involves a broad substrate scope (i–x), commercially available chemicals, has milder reaction conditions than previously reported, and requires minimal manipulation [34]. Heptamethyltropylium perchlorate (B) has been synthesized via the reaction of 1,2,3,4,5,6,7-heptamethyltropilidene (3a) with phosphorus pentachloride and subsequent treatment with perchloric acid [35].
Tropylium ion salts (C) are prepared by the oxidation of cycloheptatriene (1a) with ammonium nitrate (4a) and trifluoroacetic anhydride (4b). This reaction proceeds at room temperature in chloroform or methylene chloride. The reaction completion is indicated by the red solution of tropylium trifluoroacetate salt, but this salt cannot be isolated, so it is converted to other stable salts by the addition of suitable acids in the reaction mixture in the presence of ethanol as a solvent [36]. In the presence of acetonitrile, a hydride exchange reaction between cycloheptatriene (1a) and tritylium salts (5a) produced tropylium salt (1c or 1d) [37]. Tropylium ions can also be synthesized by the electro-oxidation of tropilidene, the “parent” hydrocarbon (1a). The electrolysis medium used was acetonitrile. In the same potential region, ditropyl (6a) is oxidized to the tropylium ion (1b) (Figure 1) [38].
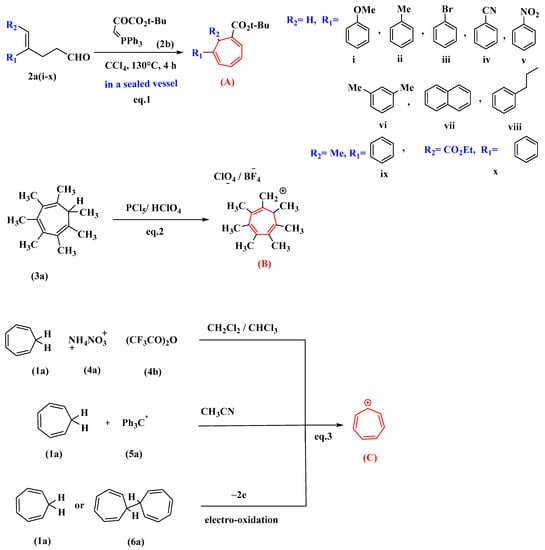
Figure 1. Various methods for synthesizing tropylium salt (C) and its derivatives (A,B). Di and trisubstituted cycloheptatrienes (A) synthesized from pyruvic ester (2a) is shown in eq.1. Heptasubstituted cycloheptatriene (B) synthesized from heptamethyltropilidene (3a) is shown in eq.2. Tropylium salts (C) synthesized from cycloheptatriene (1a) is shown in eq.3.
1.1. Spectral Studies
The 1H-NMR spectrum of the tropylium ion contains only one peak, showing that all seven protons are equivalent. Similarly, 13C-NMR of the tropylium ion also shows one peak, indicating that all seven carbons are equivalent (Figure 2). The ″C, ′H spin–spin coupling constants for tropylium fluoroborate were measured from the 13C-NMR spectra of tropylium ions using a distinct experimental technique comprising highly deuterated compounds and ′D-decoupling [39]. The coupling constants for the tropylium ion were found to be ′J = 166.79, ′J-0, ″J = 9.99, and ″J = −0.64 Hz [40].
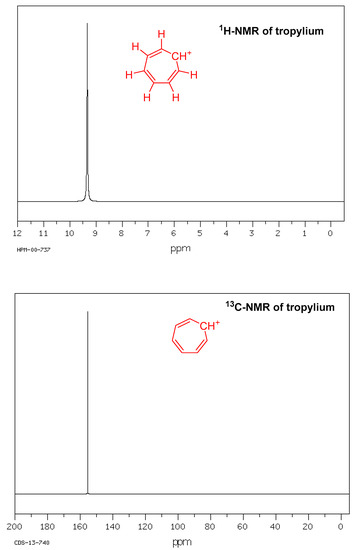
Figure 2. 1H-NMR and 13C-NMR of tropylium ion.
The tropylium ion’s UV spectrum can only be obtained in water at low pH. The specrum is analogous to that of 7-hydroxycycloheptatriene (tropyl alcohol) at high pH and mixtures at intermediate acidity. The tropylium ion has a λmin of 247 nm (log e 3.60) and a λmax of 275 nm (log e 3.64). Only four bands of reasonable intensity could be seen in the infrared spectra of tropylium bromide [41]. Compared to the spectra of the isomeric bromo- and chlorotropilidenes, which are more complex and exhibit at least twelve bands of moderate intensity, this simplicity is more compatible with the high symmetry anticipated for the tropylium ion [42].
The tropylium cation is a common fragment in the mass spectra of molecules containing benzyl units. The characteristic signal appears at m/z = 91. It represents the common benzyl fragment (PhCH2+), which, upon rearrangement, forms the highly stable tropylium cation (C7H7+). For example, the molecular ion peak of ethyl benzene appears at m/z = 106, which, upon ionization, undergoes benzylic cleavage to give the tropylium ion base peak at m/z = 91 (Figure 3).
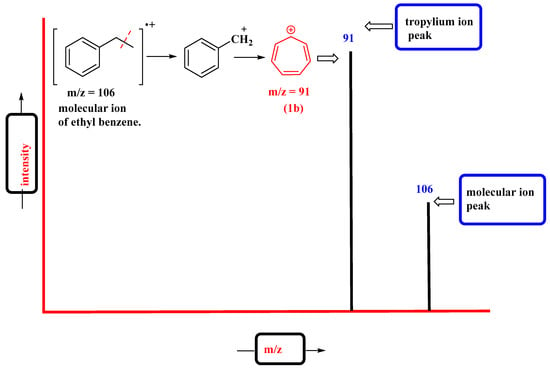
Figure 3. Mass spectrum of ethyl benzene representing characteristic tropylium ion peak.
The spectra of [M + H]+ ions of 1,3,5-cycloheptatriene and various 7-alkyl-1,3,5- cycloheptatrienes were measured after gas-phase protonation [43]. Ring contraction of the dihydrotropylium ions to protonated toluene (toluenium ions) and protonated ethyl benzene (ethyl benzenium ions) before fragmentation [44] is indicated by the loss of CH4 from the parent ion [1 + H]+ and the virtually exclusive loss of C2H4 from the methyl derivative [3 + H]+. Ions [3+ H]+ isomerize to xylenium ions as protonation becomes more exothermic [45].
According to 1H-NMR studies, (1a) is 30% as aromatic as benzene, and tropylium is 22–50% as aromatic as benzene [46]. The annelated dihydropyrenes (tropone-fused and 1,3,5-cycloheptatriene-fused) were synthesized to achieve these estimations (tropylium-fused). The tropylium cation was analyzed after treatment with HBF4 [47]. The isomeric cycloheptatrienes (70% yield) were produced when the ketone was reduced with AlH3 (generated from AlCl3/LiAlH4) in ether/benzene at 25 °C [48].
1.2. Reactivity of Tropylium Ion
The tropylium cation acts as a Lewis acid in the base, water, and is in equilibrium with the covalently bonded carbinol and the hydronium ion (Figure 4). When water is used as the reference base, the tropylium ion has a value of K = 1.8 × 10−5, which implies that it is nearly as acidic as acetic acid.
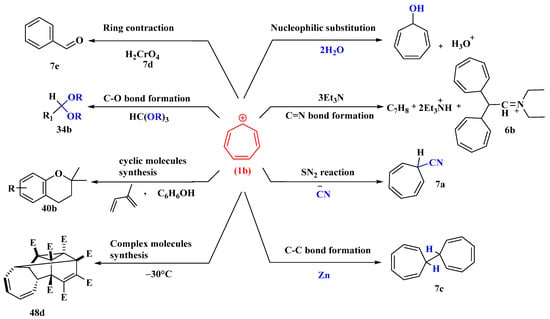
Figure 4. Important reactions of tropylium ion.
To make substituted cycloheptatrienes, the tropylium cation was treated with the hindered bases triphenylmethyl sodium, 2,6-dimethoxyphenyl lithium, and 2,4,6-tri-t-butyl phenyl lithium. Where feasible, it reacts with tertiary amines to produce ammonium salts, which can then be hydrolyzed to form tropylated aldehydes and ketones (Figure 4). When it reacts with trimethylamine, it yields a quaternary ammonium salt, which decomposes into trimethylammonium fluoroborate and a mixture of tropone and ditropyl ether when exposed to the air [49].
The tropylium ion, which can be easily prepared, reacts as an electrophilic reagent with bases such as pyridine (C5H5N), triethylamine (NEt3), and dimethyl sulfoxide (DMSO) to form adducts; with water, hydrogen sulfide, and ammonia to give ditropyl ether, sulfide, and amine, respectively; with the bases acetamide, benzamide, and succinamide to form the N-tropyl derivatives; and with cyanide ions to give tropyl cyanide (7a), which can be hydrolyzed to an amide (7b) and reacts with phenyl magnesium bromide to give desoxybenzoin. The tropylium ion is reduced by zinc dust to ditropyl (7c) [50].
Tropyl alcohol is generated first by the interaction of tropylium ions with water, and since it is in equilibrium with the tropylium ions, it reacts further to give the less soluble ether. Ditropyl sulfide is formed when hydrogen sulfide and aqueous tropylium bromide react rapidly. Tropylium bromide reacts similarly to saturated aqueous ammonia to produce ditropylamine, a secondary amine. The primary result of its reaction with ethereal ammonia is tritropylamine, a tertiary amine. Dimethylamine normally reacts with tropylium bromide to form dimethyltropylamine. Tropylium bromide interacts smoothly with aqueous potassium cyanide, giving a liquid product, C7H7N, in excellent yield. The tropylium ion can be oxidized either by chromic acid (7d) in acetic acid or by silver oxide in water to afford benzaldehyde (7e) in good yield [51] (Figure 4).
Potassium heptaphenylcycloheptatrienide is formed by reducing heptaphenyltropylium bromide with potassium (Figure 5). The anion reacts with the tropylium ion to form the heptaphenylcycloheptatrienyl radical, whereas reduction by potassium fragments the anion into stilbene and the pentaphenylcyclopentadienyl anion [52].
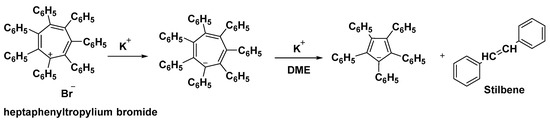
Figure 5. Reduction of heptaphenyl tropylium bromide in presence of potassium ion.
Sargent and his colleagues discovered that the 7-cycloheptatrienyl group, presumably through its nor-cardienyl valence tautomer, is incredibly effective at stabilizing adjacent cationic carbon atoms. Diazotization of the amine under various circumstances produced the most profound results. Two organic compounds were produced when treated with an equivalent of nitrosonium fluoroborate in acetonitrile at room temperature. Heptafulvene was shown to be the most important [53].
It was reported that the generation, trapping, and evaluation of a dehydrotropylium ion resulted in a reduced level of aromatic stabilization. The Jones group produced η2-platinum (0), palladium(0), and zirconium(II) complexes of a dehydrotropylium ion or similar cations that exhibit no to moderate C−C bond alternation (Figure 6) [54].
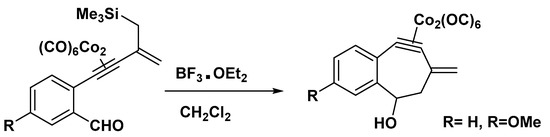
Figure 6. Preparation of a dehydrotropylium ion complex.
For a moderate Swern-type oxidation of a range of alcohols (8b), a novel dimethyl sulfoxide activation technique using 1,1-dichlorocycloheptatriene (8a) has been devised (Figure 7). With this simple and efficient technique, moderate to excellent yields of carbonyl compounds (8c) may be produced [55].
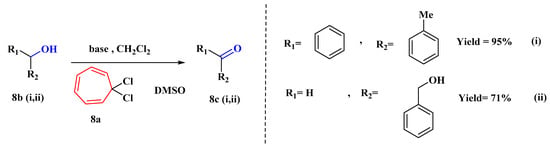
Figure 7. Activation of DMSO using 1,1-dichlorocycloheptatriene.
In the presence of palladium, the hydrogenation of heptamethyl cycloheptatrieneheptacarboxylate affords a stereoisomer of heptamethyl cyclohepteneheptacarboxylate with the cis position of five ester groups with a yield of 60%, as well as the corresponding cyclohepta-1,3-dienes as trans, trans and trans, cis isomers. After a reduction of one double bond and intramolecular cyclization, the reaction with sodium borohydride resulted in the stereoselective formation of the bicyclic molecule. The chemicals produced were analyzed using X-ray diffraction [56].
In aqueous acetic acid and other solvents, the oxidation of 1,3,5-cycloheptatriene (1a) by 4 equivalents of ceric ammonium nitrate (9b) produces benzaldehyde (9c), benzene (9d), and carbon monoxide (Figure 8). Three sets of experiments are given, all of which strongly suggest that the tropylium ion is an intermediate in this oxidation. As per the evidence given, the oxygen of the benzaldehyde generated by the ceric ammonium nitrate oxidation of (1a) in anhydrous acetonitrile originates from the nitrate ion [57].
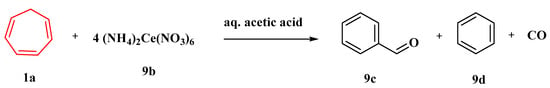
Figure 8. Oxidation of cycloheptatriene by ceric ammonium nitrate.
Dichlorocarbene preferentially adds in a 1,2-fashion to the formal “anti-Bredt”-type double bond of the aromatic ring to produce the nor-caradiene, which immediately rearranges to the bridged cycloheptatriene and then to the isomeric product through a [1,5] sigmatropic chlorine migration [58]. The thermal elimination of hydrogen bromide from dibromotropilidene (10a) produces cycloheptatrienylium bromide (10b), which is hydrogenated to cycloheptane (10c). Tropylium chloride, which is considerably more deliquescent than the bromide, is produced by dissolving tropylium bromide in ethanol, passing it through hydrogen chloride, and precipitating it with ether (Figure 9) [59].
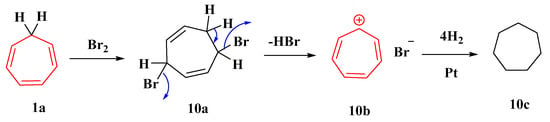
Figure 9. Elimination of HBr from dibromotropilidine.
2. Nucleophilic Substitution Reactions
The oxidizing potential of the tropylium ion to bring about the alpha-cyanation of amines was reported by Lambert [60]. Potassium cyanide (KCN) was used to synthesize aminonitriles, with tropylium being used as a catalyst, as shown in Figure 10. Thirteen substrates were reported, including 17-β-cyanosparteine synthesis (gram scale) and oxidative Aza-Cope rearrangement. The action of the tropylium ion on an amine substrate was investigated. The synthesis of the iminium ion (11b) was facilitated in the absence of KCN, but with KCN, it allows the tropylium ion (1c) to oxidize the amine substrate (11a) and produce alpha-cyanated product (11c) in 81% yield. It has a wide range and exhibits regioselective affinity. A high yield (90%) of α-cyanate-sparteine was also found. Substrates containing solely benzylic hydrogens or aliphatic positions that are not reactive caused complications [60].
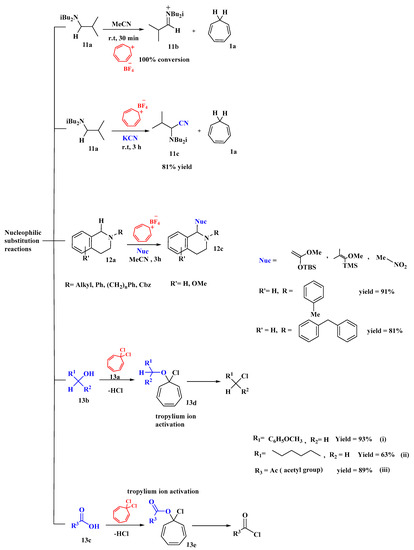
Figure 10. Tropylium as catalyst accelerated nucleophilic substitution reactions.
Nguyen created a new robust method for the oxidative functionalization of tetrahydroiso-quinolines (THIQs) at the C1 position using tropylium salts, which has a wide range of applications in medicinal chemistry [61]. He worked on N-substituted THIQs with multiple nucleophiles, such as Grignard and nitromethane, and synthesized alkylated and arylated THIQs with solid results through iminium intermediates. These iminium intermediates were generated with silyl enol ethers, which gave quick access to THIQs in good to high yields (12c). Furthermore, the alkylation of substrates (12a) with nitromethane (i) gave products in good yields (12c). These tropylium-based processes involved hydride abstraction, accompanied by the nucleophilic quenching of intermediates, and generated cycloheptatriene as a byproduct [61].
A technique for the activation of alcohols using tropylium cations for dehydrative nucleophilic chlorination and bromination was reported by Nguyen et al. [62]. In addition to the activation of alcohols, the researchers proposed that the tropylium cation could also be used to activate carboxylic acids for nucleophilic acyl substitution in the same manner. The reagent 1,1-dichlorocycloheptatriene (13a) can be generated in situ or assembled from readily available starting materials and stored under inert conditions for an extended period. The reaction of (13a) with alcohols (13b) and acid substrates (13c) converted them to chlorides and acid chlorides, respectively. The reactivity decreased from an activated primary alcohol to a non-activated primary alcohol and then to a non-activated secondary alcohol. The reaction proceeded via the formation of tropylium alkoxy (13d) and (13e) and electron-donating and electron-withdrawing groups did not affect the reactivity. The conversion of carboxylic acids (13e) to acid chlorides resulted in high yields, but non-activated primary and secondary alcohols (13b) required elevated conditions to work and resulted in lower yields (Figure 10). Moreover, it was found that this method of conversion was easy, rapid, and clean, proceeded under mild conditions, and required a short reaction time [62].
3. C-N Bond Formation Reactions
Research based on the reaction of monocarboxylic acid hydrazides (14a) with tropylium salts was performed by the Yunnikova research group [63]. They combined three distinct substrates (nicotinic acid hydrazide, isonicotinic acid hydrazide, and furan-2-carbohydrazide) with two different tropylium salts, perchlorate (1d) and tetrafluoroborate (1c), to create biologically significant products. The author’s work was praised since the technique was straightforward, the reactions were carried out in water at room temperature, and good yields were obtained. Using tropylium perchlorate (1d), the product N′, N′-di(cyclohepta-2,4,6-trien-1-yl)pyridine-4-carbohydrazide (14b) was produced with the highest yield (93%) (Figure 11). The products were further clarified based on NMR and mass spectrometry (MS) studies [63].
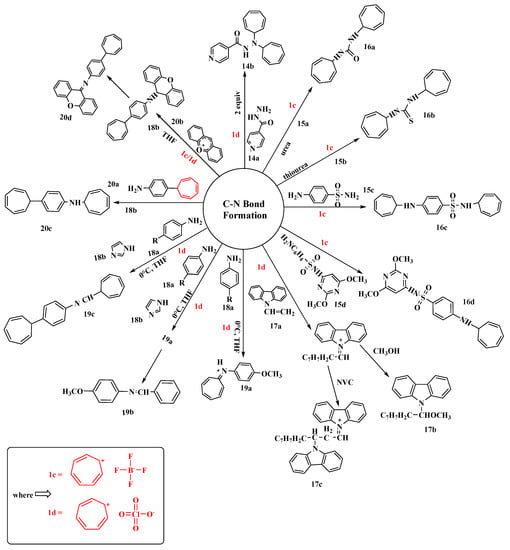
Figure 11. Tropylium-mediated synthesis of compounds containing C-N bonds.
By reacting urea (15a), thiourea (15b), sulfonamide (15c), or sulfadimethoxine (15d) with (1c), novel compounds were produced by substituting a hydrogen atom with tropylium at the amide nitrogen or the amino group of the benzene ring. Furthermore, the tropylium-salt-containing 1,3,5-cycloheptatriene was quite stable and possessed unique pharmacological properties. The primary goal of this research was to apply tropylation reactions to the graph of a well-known antimicrobial compound. Compounds such as N,N′-di(cyclohepta-2,4,6-trien-1yl)urea (16a), N,N′-di(cyclohepta-2,4,6-trien-1-yl)thiourea (16b), N-(cyclohepta-2,4,6-trien-1-yl)-4-(cyclohepta-2′,4′,6′-trien-1-yl-amino)benzenesulfonamide (16c), and sulfadimethoxine [4-amino-N-(2,6-dimethoxy-4-pyrimidinyl)benzenesulfonamide] (16d) have been synthesized [64].
Bowyer, along with his colleagues, reported the cationic polymerization of N-vinyl carbazole (17a) (NVC) initiated by tropylium hexachloroantimonate and tropylium perchlorate (1d) (Figure 11) [65]. The adiabatic calorimetric technique was used to determine the rates of reaction, and the order of the half-life was 1–2 s. The initiation reaction of the tropylium ion with a monomer occurs readily with ion pairs (low concentration), whereas propagation occurs through free cations. To start the process, stable carbonium ion salts were employed. It was observed that the polymer (17c) was formed in high yield through a charge-transfer complex when the two reagents (17a) and (1d) reacted at a temperature above −50 degrees. The molecular weight of the polymer produced using perchlorate catalysis was found to be lower than that of the polymer created via hexachloroantimonate catalysis. Conductance measurements were utilized to investigate the tropylium salt ion-pair dissociation equilibria [65].
The tropylation of arylamines using tropylium perchlorate (1d) was studied by Yunnikova [66]. She was able to synthesize 4-(7-cyclohepta-1,3,5-trienyl)-N-(1-cyclohepta-2,4,6-trienyl)aniline by the tropylation of p-anisidine with (1d), which formed the salt 8-p-methoxyphenyl-8-azaheptafulvenium perchlorate (19a) in 72% yield. Because of the tropylium ring contraction in the presence of imidazole, this product was transformed into N-benzylidene-4-methoxyaniline (19b). The yield of (19b) was a bit higher (76%) than that of (19a) due to the presence of imidazole (18b) as an activator. Moreover, replacing the methoxy in p-anisidine with tropylium altered the reaction’s path and produced (19c) in a yield 56% less than the previous compounds synthesized [66].
Yunnikova’s research group proposed that tropylium, xanthylium, and tritylium salts have various stabilities, which affect how they react with biologically active amines [67]. The reaction of (1d) and (1c) with 4-(cyclohepta-2,4,6-trien-1-yl) aniline (20a), followed by the hydrolysis of the N-(cyclohepta-2,4,6-trien-1-yl) derivative, is responsible for the difference in their relativities. Tritylium perchlorate failed to react with (20a), whereas the N-xanthenyl derivative was dehydrogenated. The reactions of pyrimidin-2-amine with tropylium, xanthylium, and tritylium salts produced large yields with the replacement of one hydrogen atom in the amino group (Figure 8). In the synthesis of (20c), the tropylium ring remained intact with a good yield, but when using two equivalents of (20b), the yield increased from 44% to 87%. The purpose of this work was to investigate amine reactions with tropylium, xanthylium, and tritylium salts and estimate the stability and yields of the products. 1H-NMR and mass spectra, as well as the X-ray analysis of amines, were used to confirm the structure of all isolated compounds [67].
4. C-C Bond Formation Reactions
Nguyen’s research group successfully created a series of new cationic organic dyes from widely accessible synthetic precursors utilizing the tropylium ion as an electron-withdrawing chromophore in a simple one-pot chemical method [68]. The synthesis was carried out by mixing aniline (21a) and two equivalents of (1c) to produce (21b) and (21c) in good to excellent yields. The reaction proceeded with electrophilic aromatic substitution with the first equivalent of (1c) and by hydride abstraction with the second equivalent. By controlling the reaction conditions, the intermediate (21b) could be isolated (Figure 12). These tropylium-based organic dyes were stable and water-soluble, with high visual absorption, as demonstrated by easy and practical synthesis methods. All of the organic dyes mentioned in this researchticle were pH-responsive and exhibited unique physicochemical properties, such as solvatochromism, redox-responsivity, Lewis basicity, and fluoride anion sensitivity; however, the researchers only looked at two members of this family, Trop-26DIPA (diisopropylamine) and Trop-DEA (diethanolamine) [68].
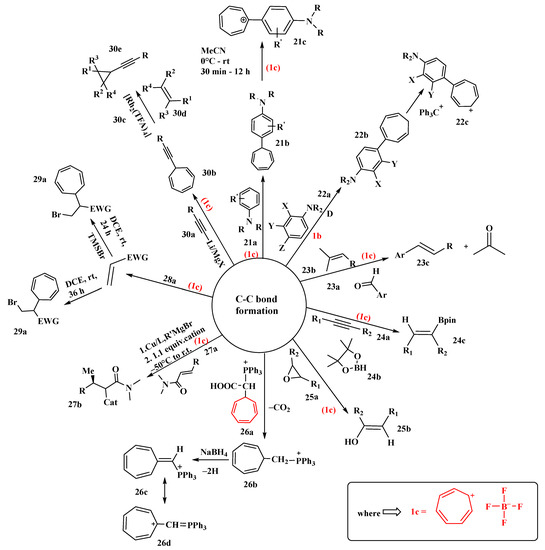
Figure 12. Tropylium-promoted synthesis containing C-C bond formation.
Looker proposed the use of tropylium tetrafluoroborate (1c) in the alkylation of aromatic rings such as 1-dimethylaminonaphthalene, N, N-dimethylaniline, and N, N-diethyl-m-toluidine (22a), activated by dialkylamino groups [69]. NMR studies revealed two sets of peaks, indicating that the products obtained were isomeric mixtures of dialkylaminoarylcycloheptatrienes (22b) formed via double-bond shifts in seven-membered rings. Some of these isomers were isolated in yields of 80–84%. The thermal isomerization of 7-substituted 1,3,5-cycloheptatriene was examined by NMR studies. Triphenylmethyl fluoroborate was used to oxidize the substituted cycloheptatriene to the corresponding tropylium salt alkylating agent (22c), generating deep-blue, stable compounds (Figure 12) [69].
Based on preliminary studies of olefin–olefin metathesis reactions, the Nguyen research group investigated the carbonyl olefin metathesis reaction [70]. In these reactions, they illustrated the use of tropylium as an organocatalyst. Tropylium catalysts were used in intramolecular, intermolecular, and ring-opening carbonyl olefin metathesis (COM) reactions in their research. They started their investigation with intramolecular COM reactions using different aldehydes (23a) and isopropylidine-bearing olefin substrates (23b). The final product formed was a trans-isomer (23c) with a good yield. It was further proved that electron-deficient aryl aldehydes did not lead to any productive outcomes. Tropylium ions preferred both inter- and intramolecular interactions, but with intramolecular reactions, a 93% yield was achieved. Catalysts such as iron chloride and tritylium ions, which were previously employed, favored only one type [70].
A specific method for efficiently promoting the hydroboration reaction of alkynes (24a) and epoxides (25a) by utilizing tropylium salts, which are often used as organic oxidants and Lewis acids, is described. The tropylium ion (1b) may abstract a hydride from borane reagents (24b) and use it to activate the reaction by forming a borenium ion. The catalytic activity of (1c) in the synthesis of phenylacetylene (25b) and pinacolborane (24c) was studied. The fact that potassium or tetrabutylammonium salts of the same counterion either had no catalytic activity or hindered the reaction emphasized tropylium’s importance. It is noteworthy to mention the authors whose study indicated that tropylium salts function as efficient reaction promoters and that hydride abstraction of pinacol borane with the tropylium ion initiated several intriguing pathways of reactions. A variety of substrates were tested, and it was shown that a satisfactory yield could be obtained with a 5% mole tropylium catalyst and a 12 h reaction time. However, with epoxide substrates (25a), a high catalytic amount and a longer reaction time are needed to give the completely hydrolyzed product (25b) [71].
Triphenylphosphonium tetrafluoroborates were synthesized by Cavicchio et al. [72]. They examined the possibility of synthesizing onium salts and ylides due to the possible synthetic utility in the realm of cycloheptatriene and tropylium ion derivatives. They discovered in preliminary research that the simple direct route to phosphonium salts of (26a) through the interaction of the tropylium ion (1b) with phosphonium ylides is confined to stabilized ylides from (26c) but fails when R = H (26b) due to the extremely high reactivity of methylenetriphenylphosphorane (Figure 12). The authors were successful in explaining the stabilizing effect of the phosphonium group on the heptafulvene system, comparable to cyano or carbonyl groups. The synthesis of (26c) was reported, which was then successfully converted to (26d) [72].
Sebesta studied different techniques for trapping chiral enolates (ideally silyl enolates) generated by Lewis acids, which are synthesized by the enantioselective conjugate addition of Grignard reagents to unreactive Michael acceptors [73] (Figure 12). This one-pot reaction was accomplished by the use of different electrophilic reagents. The trapping of Mg-enolates by carbocations was reported. The tropylium cation was the first electrophile to be utilized. The investigation started with amides (27a) that led to functionalized molecules (27b). The yield was determined by changing the solvents, reagent side chains, and electrophiles. The polar additive 1,3-dimethylimidazolidin-2-one (DMEU) was added, as it afforded the highest conversion. It was discovered experimentally that steric parameters play a significant role in the reaction outcome and that trapping reactions are highly dependent on the electrophile structure. A rapid decrease in the yield was observed as the R group became bulkier (27a). The overall yields of all trapping reactions varied from 15 to 65%, and it was assumed that the low yields were due to silyl enolate’s poor reactivity. It is worth mentioning the author, whose study revealed that numerous functionalized products may be produced using a single-pot approach that was previously unattainable using other methods [73].
Nguyen developed a new method using halide ions for activating electron-deficient olefins for nucleophilic attack on stabilized carbenium ions in a catalyst-free, metal-free, non-radical fashion [74]. This new chemical reaction thus allows the direct carbohalogenation of electron-deficient olefins. He discovered that tropylium bromide interacted with electron-deficient alkenes (28a) such as methyl acrylate in a highly regioselective manner to form the racemic bromocyclohepta-trienylated compound (29a). The substrates with C-C multiple bonds without acceptor substituents such as amide groups gave high yields up to 91%. He also described transformations that allowed easy access to a diverse variety of synthetically valuable substituted cycloheptatrienes. The author further revealed that electron-deficient alkynes behaved similarly to their olefinic analogs [74].
A two-step alkynylcarbene transfer reaction for the assembly of alkynyl cyclopropanes was developed using commercially available tropylium tetrafluoroborate, terminal alkynes, and alkenes. The reaction proceeded through the decarbenation of readily available 7-alkynyl cycloheptatrienes (30b) (prepared in one step from commercially available terminal alkynes (30a) and (1c)). This reaction was catalyzed by rhodium (II), which outmaneuvers the fundamental problem associated with 1,6-enyne-containing substrates with Lewis acids, which usually evolve via 6-endo-dig cyclization or ring-contraction pathways under metal catalysis. Based on this discovery, the researchers were able to synthesize a wide variety of cis-alkynylcyclopropanes (30e) bearing C (sp3−), C (sp2)-, and C (sp)-, H-, Si-, or Ge substituents in the alkyne terminus (Figure 12). It was observed that (30b) with bulky alkyl groups and phenyl groups with electron-donating substituents led to high yields of (30e). Moreover, less activated alkenes and electron-rich alkenes (30d) were both compatible with the reaction conditions (Figure 12). The formation of rhodium (II) alkynylcarbene intermediates, which react smoothly with alkenes (30d) to give cyclopropanes, was also supported by experimental and theoretical investigations [75].
5. Ring-Contraction Reactions
The relevance of the ring contraction of cycloheptatriene is highlighted by Alsamarrai [76]. The interaction of tropylium fluoroborate (1c) and arsonium ylide (31d) in dichloromethane produced 3-phenyl-1,2-dicarbomethoxyprop-2-enylidentriphenyl arsonium salt (31e). The arsonium ylide was synthesized by the reaction of (31b) and (31a) dimethyl acetylenedicarboxylate (DMAD). The product (31e) underwent further treatment with aqueous NaOH to obtain the Z-isomer arsorane (31f) (a colorless gum, yield = 77.7%) (Figure 13). It involved the cycloheptatriene ring contraction of the salt (31e). The Z-isomer (31f) then isomerized to the E-isomer (31g) under sunlight irradiation and was extremely important. The relationship between the two isomers was determined to be geometrical isomerism by utilizing analytical HPLC. An alternate approach to the isomeric product (31f) was also devised by directly treating (31c) with (1c) in acetonitrile, which proved to be a more efficient procedure. The reactivity of isomer (31f) with (1c) was further investigated [76].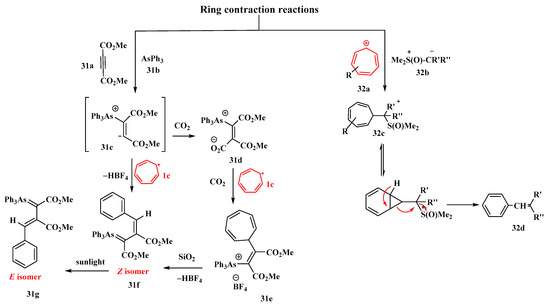
Figure 13. Role of tropylium in ring-contraction reactions.