Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Monica Lopez-Guerra and Version 2 by Rita Xu.
Major advances in the understanding of acute myeloid leukemia (AML) pathogenesis, together with technological progress, have led us into a new era in the diagnosis and follow-up of patients with AML. A combination of immunophenotyping, cytogenetic and molecular studies are required for AML diagnosis, including the use of next-generation sequencing (NGS) gene panels to screen all genetic alterations with diagnostic, prognostic and/or therapeutic value.
- AML
- diagnosis
- flow cytometry
1. Introduction
Acute myeloid leukemia (AML) is characterized by a high biological heterogeneity both at diagnosis and during disease evolution. An exhaustive characterization of immunophenotypic and molecular profiles is required to guide the clinical management of AML patients. In recent years, growing evidence has shown the prominent role of cytogenetic and mutational data in AML and, accordingly, the recently updated World Health Organization (WHO) [1] and International Consensus Classification (ICC) [2] classifications of myeloid neoplasms as well as the European LeukemiaNet (ELN) recommendations [3] have incorporated genetics into their diagnostic, prognostic and therapeutic algorithms. In this scenario, a combination of immunophenotyping and genetic studies are required for AML diagnosis and follow-up. Measurable residual disease (MRD) has strong prognostic and predictive value in AML, and its accurate monitoring is crucial for disease management. The plethora of aberrant phenotypic and molecular characteristics of AML cells represents a chance to track this disease but, at the same time, constitutes a major challenge for hematopathology laboratories.
2. Diagnosis
2.1. Flow Cytometry for Lineage Assessment
Multiparametric flow cytometry (MFC) consists of the recognition of cells based on antigen detection through a combination of fluorochrome-labeled monoclonal antibodies, which allows phenotypic characterization and the quantification of a specific cell population. When acute leukemia is suspected, morphologic and immunophenotypic examinations of the bone marrow and peripheral blood are the quickest techniques to confirm the diagnosis (Figure 1). The latter is also essential when assigning the leukemic lineage: a crucial step that determines further diagnostic tests and therapeutic strategies. Lineage-assigning antigens are shown in Table 1. Myeloperoxidase (MPO) expression is the hallmark of myeloid commitment, though it is not always present. AML with minimal differentiation is defined by an absence of lineage-assigning antigens in combination with the expression of at least two other myeloid-related antigens (e.g., CD13, CD33, CD117). Monocytic AML often loses MPO expression and is characterized by specific markers such as CD11c, CD14, CD64 and lysozyme.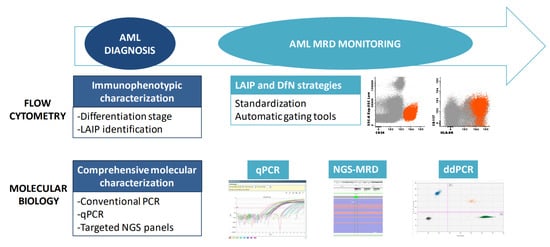
Figure 1. Tools for diagnosis and monitoring of AML. A combination of immunophenotyping and molecular studies is required for AML diagnosis and MRD monitoring. MRD: Measurable residual disease; LAIP: Leukemia-associated immunophenotype; DfN: Different from normal; qPCR: quantitative PCR; ddPCR: Droplet digital PCR.
Table 1. Flow cytometry diagnostic and MRD markers.
| Diagnostic Markers | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lineage assigning antigens | ||||||||||
| AML with Recurrent Genetic Abnormalities | AML with Defining Genetic Abnormalities | Myeloperoxidase | Myeloid lineage | |||||||
| Acute promyelocytic leukemia (APL) with t(15;17)(q24.1;q21.2)/ | PML::RARA | ≥10% | Acute promyelocytic leukemia (APL) with | PML::RARA | fusion | No threshold | CD11c, CD14, CD64, lysozyme | Myeloid lineage with monocytic differentiation | ||
| APL with other | RARA | rearrangements | ≥10% | CD19 | B-lineage. Requires also at least one (CD19 strong) or two (CD19 weak) from CD22, CD10 and CD79a | |||||
| AML with t(8;21)(q22;q22.1)/ | RUNX1::RUNX1T1 | ≥10% | AML with | RUNX1::RUNX1T1 | fusion | No threshold | CD3 (surface or cytoplasmic) | T-lineage | ||
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/ | CBFB::MYH11 | ≥10% | AML with | CBFB::MYH11 | fusion | No threshold | Myeloid differentiation-associated antigens | |||
| AML with t(9;11)(p21.3;q23.3)/ | MLLT3::KMT2A | ≥10% | AML with | KMT2A | rearrangement | No threshold | CD13, CD33, CD11b, CD15, CD64 | Myeloid | ||
| AML with other | KMT2A | rearrangements | ≥10% | CD14, CD36, CD64, CD4, CD11c | Monocytic | |||||
| AML with t(6;9)(p22.3;q34.1)/ | DEK::NUP214 | ≥10% | AML with | DEK::NUP214 | fusion | CD41, CD42b, CD61, CD36 | Megakaryocytic | |||
| No threshold | ||||||||||
| AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)/ | GATA2 | ; | MECOM(EVI1) | ≥10% | AML with | MECOM | rearrangement | No threshold | CD235a, CD71 strong, CD105, CD36 | Erythroid |
| AML with other | MECOM | rearrangements | ≥10% | CD203c, CD123 | Basophil | |||||
| AML with other rare recurring translocations | ≥10% | AML with | NUP98 | rearrangement | No threshold | CD123, CD4, HLA-DR strong, CD303, CD304 | Dendritic | |||
| AML with | RBM15::MRTFA | fusion | No threshold | CD117 strong | Mastocytic | |||||
| AML with t(9;22)(q34.1;q11.2)/ | BCR::ABL1 | ≥20% | AML with | BCR::ABL1 | fusion | ≥20% | MRD markers | |||
| AML with mutated | NPM1 | ≥10% | AML with | NPM1 | mutation | No threshold | Basic markers | |||
| AML with in-frame bZIP | CEBPA | mutations | ≥10% | AML with | CEBPA | mutation | ≥20% | CD34, CD117, HLA-DR, CD45, CD13, CD33 | Myeloid precursor identification | |
| AML and MDS/AML with mutated | TP53 | 10–19% (MDS/AML) and | CD7, CD56 | Lymphoid antigen aberrancies | ||||||
| Other useful markers | ||||||||||
| CD64, CD14, CD11b, CD4 | Monocytic | |||||||||
| CD19, CD2, CD5 | Lymphoid antigen aberrancies | |||||||||
| CD38, CD123, CD133 | Leukemia stem cell identification | |||||||||
2.2. Molecular Biology
Over the last decade, genomic studies based on Next-Generation Sequencing (NGS) have dissected the molecular profile of AML, with the description of new mutations, copy number variations and recurrent fusion genes [9][10][9,10]. The growth in molecular knowledge has prompted the update of both diagnostic and management recommendations for AML patients. The recently updated WHO, ICC and ELN classifications have undertaken a major role in genetic data and have been integrated into diagnostic and prognostic algorithms (Table 2) [1][2][3][1,2,3]. Genetic analyses currently mandatory in the evaluation of AML are conventional cytogenetics together with the screening of fusion genes and of gene mutations, whose number has significantly increased in comparison with prior classifications (Figure 1). Recommendations for molecular assessments in AML include the following genes: FLT3 (both internal tandem duplications, ITD, and tyrosine kinase domain, TKD, mutations), IDH1 and IDH2 as therapeutic targets, and NPM1, CEBPA, DDX41, TP53, ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1 and ZRSR2 as diagnostic/prognostic disease markers. Furthermore, the screening of the most prevalent gene rearrangements include PML::RARA, RUNX1::RUNX1T1, CBFB::MYH11, KMT2A rearrangements, BCR::ABL1 and others and should be performed when rapid information is needed for the clinical management of a patient, or if the morphology and/or immunophenotype are highly suggestive of the presence of a particular fusion gene [3].Table 2. Classification of AML according to ICC and WHO 2022.
| ICC 2022 | WHO 2022 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Category | Blasts Required for Diagnosis | Category | Blasts Required for Diagnosis | |||||||||||||
| ≥20% (AML) | ||||||||||||||||
| AML and MDS/AML with myelodysplasia-related gene mutations | ||||||||||||||||
| Defined by mutations in | ||||||||||||||||
| ASXL1 | ||||||||||||||||
| , | ||||||||||||||||
| BCOR | ||||||||||||||||
| , | ||||||||||||||||
| EZH2 | ||||||||||||||||
| , | RUNX1 | , | SF3B1 | , | SRSF2 | , | STAG2 | , | U2AF1 | , or | ZRSR2 | 10–19% (MDS/AML) and ≥20% (AML) |
AML, myelodysplasia-related Defined by one or more cytogenetics of molecular alterations: |
| ≥20% | |
| AML with myelodysplasia-related cytogenetic abnormalities Defined by a complex karyotype (≥3 unrelated clonal chromosomal abnormalities in the absence of other class-defining recurring genetic abnormalities), del(5q)/t(5q)/add(5q), −7/del(7q), +8, del(12p)/t(12p)/add(12p), i(17q),−17/add(17p) or del(17p), del(20q), and/or idic(X)(q13) clonal abnormalities |
10–19% (MDS/AML) and ≥20% (AML) |
|||||||||||||||
| AML with other defined genetic alterations | No threshold | |||||||||||||||
| AML without recurrent genetic abnormalities | AML without defining genetic abnormalities | |||||||||||||||
| AML not otherwise specified (NOS) | 10–19% (MDS/AML) and ≥20% (AML) |
AML defined by differentiation | ≥20% | |||||||||||||