Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Segundo Gonzalez.
Chemoimmunotherapy is an emerging treatment option for cancer that combines traditional chemotherapy with immunotherapy. This approach aims to increase the efficacy of cancer treatment by simultaneously targeting cancer cells through chemotherapy and boosting the immune system’s ability to fight cancer through immunotherapy.
- immunotherapy
- chemotherapy
- immune checkpoints
1. Chemotherapy and Immunotherapy: Friends or Foes?
Chemotherapy has been the cornerstone of cancer treatment for over 70 years. In the last decade, ICIs have revolutionized cancer treatment, becoming the frontline therapy for many cancers. In some tumors, such as melanoma, renal cell carcinoma, and others, immunotherapy has largely replaced chemotherapy owing to its clinical benefits and toxic profile, and generally being more manageable and less severe than chemotherapy and radiotherapy [1]. Nevertheless, despite this impressive clinical revolution, the rate of response to immune checkpoint blockade monotherapy is usually around 20% across solid tumors due to primary and acquired resistance to ICIs [2]. The identification of novel biomarkers to discriminate the best responders and the combination of ICIs with other therapeutic modalities are promising avenues to improve their clinical response and patient outcomes.
Cytotoxic chemotherapy has widely been regarded as immunosuppressive, since it causes dose-dependent myelosuppression, thereby suggesting an antagonistic effect with immunotherapy. Nevertheless, accumulated preclinical and clinical evidence has shown that certain chemotherapeutic drugs may act, under defined conditions, as strong adjuvants for enhancing antitumor immunity and, as a result, may potentiate immunotherapy [3]. Accordingly, more than 200 clinical trials combining PD-1/PD-L1 blockade with chemotherapy have already been completed, and several chemo-immunotherapy combinations have recently been clinically approved owing to their improvement in patient survival, with generally expected safety profiles of the known toxicities of each agent [4,5][4][5].
2. Immune Checkpoints
Immune checkpoints are crucial regulators of the activation of T cells that play a physiological role in preventing anti-self-responses and autoimmunity. In advanced cancers and chronic viral infections, chronic T cell stimulation induces and up-regulates the expression of inhibitory immune checkpoints, including Programmed Cell Death 1 (PD-1) and Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4), displaying an exhausted phenotype characterized by decreased proliferation, differentiation, and survival. T cell exhaustion limits unwanted immune responses in chronic viral infections, but hinders antitumor immunity in advanced cancers. Monoclonal antibodies targeting inhibitory immune checkpoints, including CTLA-4, PD-1, Programmed Cell Death 1 Ligand 1 (PD-L1), and Lymphocyte-Activation Gene 3 (LAG-3) capable of interfering with negative signals provided by these molecules have revolutionized cancer treatment. Despite their impressive clinical results, the rate of response to ICI monotherapies is far from being satisfactory, and a majority of patients with cancer have failed to exhibit clinical benefits from these therapies [2]. Still, decades of chemotherapeutic treatment of cancer have shown that, with rare exceptions, single drugs targeting individual steps of carcinogenesis have demonstrated limited capability to cure due to the heterogeneity and complexity of advanced cancers. Combining different drugs and therapeutic modalities is an obvious strategy to improve patient outcomes [2,6][2][6].3. The Rationale behind the Combination of Chemotherapy with Immune Checkpoint Inhibitors
Cytotoxic drugs directly kill tumor cells and/or hinder their proliferation via multiple mechanisms including inducing DNA damage, inhibiting DNA replication, and/or preventing mitosis. Chemotherapeutic drugs in monotherapy have shown, with rare exceptions, limited efficacy; however, combination chemotherapy targeting multiple steps in carcinogenesis has been found to be a more effective strategy and, hence, has been widely extended and applied for cancer treatment. Combination regimens may provide a meaningful advantage over monotherapy, by maximizing cancer elimination within the range of tolerated toxicity, targeting a broader range of tumor cells with different genetic and epigenetic abnormalities among a heterogeneous tumor population, and also limiting or slowing the development of drug resistance. Conventional chemotherapy has a cytotoxic and cytostatic effect on healthy proliferating cells, especially on hematopoietic cells, causing myelosuppression. This suggests an antagonistic effect between chemotherapy and immunotherapy. In fact, some immunosuppressive drugs used to treat autoimmune diseases or to prevent transplant rejection are chemotherapeutics. Nevertheless, mounting evidence shows that the activation of host immunity decisively contributes to the efficacy of certain cytotoxic drugs; under defined conditions, they may display an immune stimulatory effect, providing an opportunity for their combination with immunotherapy [3,7,8][3][7][8]. The rationale behind this combination lies in the fact that immunotherapy has the capability to eliminate disseminated and metastatic cancer, while it is less effective in eradicating a solid tumor mass [6]. Chemotherapy may potentiate the efficacy of immunotherapy because it has the ability to debulk the primary tumor mass, decreasing the number of cells that should therefore need to be eliminated by immune cells, and also reducing the immunosuppressive factors produced by cancer cells. Additionally, certain chemotherapeutic drugs may directly stimulate antitumor immunity, which may be particularly relevant in “cold” tumors with low effector T cell infiltration within the tumor mass.4. Chemotherapy May Boost Antitumor Immunity
Abundant preclinical evidence demonstrates that the efficacy of certain chemotherapeutic agents is higher in immunocompetent mice than in their immunodeficient counterparts [9]. In good agreement, diverse studies have reported that common chemotherapeutic drugs may induce, in a dose- and schedule-dependent manner, antitumor immunity, mainly through the activation of effector T cells and NK cells and by specifically targeting the immunosuppressive tumor microenvironment (TME). In this section, the main immunomodulatory mechanisms underlying the action of chemotherapy will be discussed (Figure 1).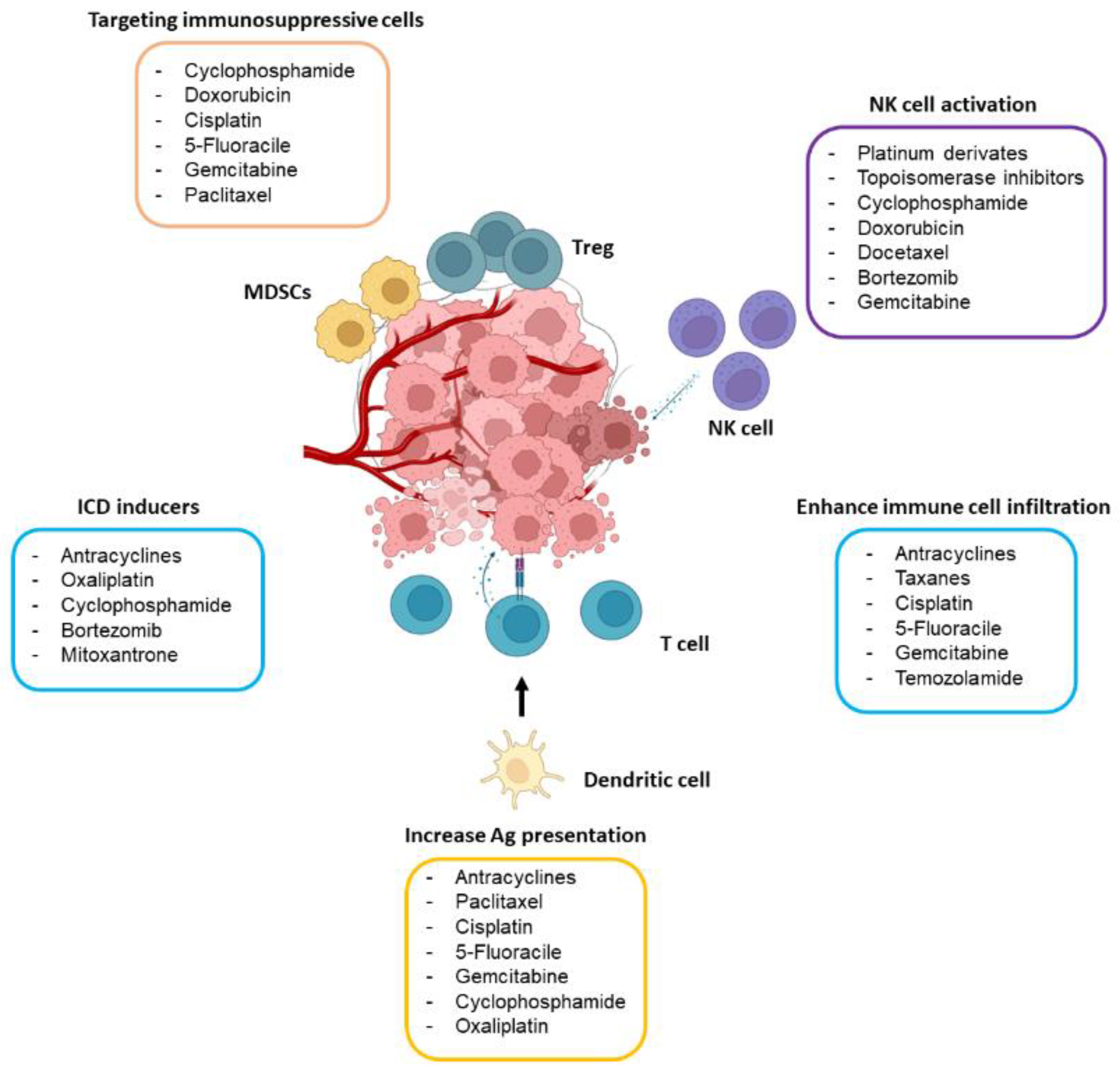
Figure 1. Main immunomodulatory effects of chemotherapeutic drugs. The drugs included in the figure may boost antitumor immunity by targeting immunosuppressive immune cells (mostly Tregs and MDSCs), activating NK cells, causing ICD, and stimulating antigen (Ag) presentation through dendritic cells and T cell activity. The dose of the drug seems to play a crucial role in its capability to stimulate the immune system.
4.1. Chemotherapy Activates T Cell Response
The type of cell death caused by cytotoxic chemotherapy is a determinant factor for triggering immunity or immune tolerance. Immunogenic cell death (ICD) is a modality of regulated cell death that results in cytotoxic lymphocytes (CTL)-mediated responses against antigens expressed by dying cells, ultimately triggering immunological memory (Figure 2) [3,7,8][3][7][8]. ICD is elicited by several cancer therapies, including radiotherapy and some chemotherapeutic drugs, such as anthracyclines, taxanes, cyclophosphamide, bortezomib, crizotinib, oxaliplatin, and other platinum-derivates (however, cisplatin is not a bona fide ICD-inducer). ICD is a potent endogenous immune adjuvant to the host innate immune system through the exposure and release of danger-associated molecular patterns (DAMPs) into the TME that are recognized by pattern recognition receptors expressed by antigen-presenting cells, mostly dendritic cells (DCs). Some DAMPs, including adenosine triphosphate (ATP) and annexin 1, enable the recruitment and chemotaxis of DCs; others, such as calreticulin, are exposed on the cell membrane acting as an “eat me signal” for the engulfment of the dying cell by DCs [8]. The release of high-mobility group protein B1 (HMGB1) and the secretion of multiple cytokines, including type I interferons, culminates in the maturation of the DCs and the recruitment and activation of the CD8 T cell-mediated immune response against the tumor cells [8]. This cascade of events promotes immune cell infiltration, shifting the tumors from “cold” to “hot” phenotypes [10].
Figure 2. Immunogenic cell death (ICD). Chemotherapeutic drugs may induce the immunogenic death of tumor cells, which results in a CD8 T cell-mediated response against tumor antigens expressed by the dying cells. ICD leads to the exposure and release of DAMPs into TME, which are mainly recognized by DCs. Some DAMPs, including ATP and annexin 1 (ANXA1), induce the recruitment of DCs; others, such as calreticulin, are expressed on the membrane of tumor cells acting as an “eat-me signal” enabling their uptake by DCs. The release of HGMB1, type I interferons, and several cytokines and chemokines culminate in the maturation of DCs and the recruitment and activation of antitumor CD8 T cells that mediate the response against the tumor and generate long-term immune memory.
4.2. Chemotherapy Dampens the Immunosuppressive Tumor Microenvironment
Advanced cancers progressively accumulate immunosuppressive cells in their TME, mostly regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs) that remain a major barrier hindering effective antitumor immunity. Low doses of several cytotoxic drugs can selectively deplete both circulating and tumor-infiltrating Tregs concomitantly, stimulating antitumor immunity (Figure 2). Interestingly, this effect is particularly well documented with low-dose cyclophosphamide [28]. Mechanistically, Tregs lack the expression of cyclophosphamide-extruding transporter ABCB1, being more sensitive to cyclophosphamide than effector immune cells [29]. It is worth mentioning that cyclophosphamide at a higher dose also induces ICD. Multiple chemotherapeutic agents, including cyclophosphamide, cisplatin, paclitaxel, 5-fluorouracil, gemcitabine, and doxorubicin, selectively eliminate MDSCs in multiple mouse tumor models, resulting in immune recovery and tumor regression [30,31,32,33,34][30][31][32][33][34]. However, recent reports suggest that, under certain conditions, chemotherapy might also induce the accumulation of MDSCs in TME [35,36,37][35][36][37]. For example, certain cytotoxic drugs such as cyclophosphamide and melphalan may cause an increase in MDSC infiltration due to the inflammatory response triggered by chemotherapy [36]. Thus, the effects of chemotherapy on MDSCs can vary depending on several factors, including the chemotherapeutic agent, dosage, and timing. Nevertheless, the clinical relevance of this preclinical evidence in cancer patients remains to be established [38].4.3. Chemotherapy Activates NK Cells
NK cells are cytotoxic innate immune cells that play a relevant role in cancer immunosurveillance and immunotherapy, particularly in hematological cancers and metastasis [39]. NK cells can eliminate malignant tumors in a non-MHC and non-tumor antigen-restricted manner through an array of activating (i.e., NKG2D, DNAM-1, NCRs) and inhibitory receptors (i.e., KIRs, NKG2A-CD94) that detect changes in the expression of their ligands during viral infection and malignant transformation. Mounting preclinical evidence shows that the DNA damage response pathway initiated by ATM, ATR, and p53, induced by multiple genotoxic drugs, triggers tumor cells to express ligands for the NKG2D receptor. This upregulation promotes NK cell-mediated cytotoxicity and IFN-γ release, which subsequently favors the upregulation of MHC class I molecules on tumor cells, sensitizing them to CTLs (Figure 1) [40]. Similarly, hyperdiploid-inducing chemotherapeutic agents, including cytochalasin D, nocodazole, and docetaxel, strongly upregulate the tumor expression of NKG2D and DNAM-1 ligands, rendering tumor cells more susceptible to NK cell-mediated lysis [41]. In patients with lung cancer, low-dose gemcitabine enhanced NK cell-mediated cytotoxicity [42], and a maintained administration of low-dose cyclophosphamide, referred to as metronomic dose (see Glossary), enhanced NK cell activity in end-stage cancer patients [43]. PD-1 is not expressed in peripheral blood NK cells from most healthy individuals; however, in the context of cancer, its expression is induced in peripheral and tumor-derived NK cells, dampening antitumor immunity, which has been correlated with poor prognosis in multiple cancer patients [44,45][44][45]. Interestingly, the response to PD-1 blockade may be enhanced by the increased number and activation of NK cells, thereby improving the clinical effectiveness, particularly in MHC class I-defective tumors [46,47,48,49][46][47][48][49]. It is worth mentioning that some tumor cells can induce PD-L1 expression on NK cells via AKT signaling, and the PD-L1 blockade results in enhanced NK cell activity and tumor regression [49]. This provides a potential explanation as to why some patients lacking PD-L1 expression in cancer cells still respond to anti-PD-L1 therapy. Collectively, accumulating evidence suggests a relevant contribution of NK cells to the clinical success of ICIs and, in this scenario, chemotherapy may improve their effectiveness through the activation of this immune subset.5. Determinants of the Success of Chemo-Immunotherapy
5.1. The Right Dose of Chemotherapy
Chemotherapy drugs cause dose-dependent myelosuppression and, in the clinic, are usually administered at the maximum tolerated dose causing immunosuppression. Despite variable clinical results, metronomic chemotherapy is a promising alternative to the conventional dosage that may have a beneficial effect on TME by inhibiting tumor angiogenesis and boosting antitumor immunity, while avoiding toxicity caused by maximum-tolerated dose treatments [50]. The underlying mechanism is far from being elucidated, but maximum tolerated dose regimens are associated with a depletion of effector immune cells, including CD4 and CD8 T cells, NK cells, and γδT cells, whereas low-dose regimens selectively target immunosuppressive Tregs and MDSCs, ameliorate T cell exhaustion, promote the maturation and activation of DCs, and concomitantly activate the NK and T cell-mediated antitumor immunity [33,51,52,53,54,55,56][33][51][52][53][54][55][56]. Standard regimens, but not metronomic doses of temozolomide or paclitaxel, have abrogated the survival advantage provided by a PD-1 blockade in murine glioma and TNBC models, respectively [53]. Metronomic gemcitabine in models of non-small-cell lung carcinoma (NSCLC) and low-dose cyclophosphamide in neuroblastoma have led to the increased efficacy and diminished toxicity of the PD-1 blockade due to reduced tumor angiogenesis dampening Tregs and enhancing the T cell effector response [57]. Along these lines, metronomic oxaliplatin and pemetrexed together with a PD-1 blockade have successfully activated T cell immunity, eliciting tumor-specific long-term immune memory in colon cancer models [58]. Similar results have been reported for combined metronomic chemotherapy with a multi-peptide vaccine and anti-PD-1 checkpoint inhibition in melanoma in vivo [59]. These preclinical data suggest that the balance between active antitumor immunity and tumor elimination with less toxicity could be critical for the success of chemo-immunotherapy. In clinical settings, chemotherapy is conventionally administered at a maximum tolerated dose, and the effect of metronomic chemotherapy has not yet been well-established [28,60][28][60]. This is particularly true for older patients, who are under-represented in current standardized clinical trials, and in whom a metronomic dose may ameliorate its adverse effects.5.2. The Timing of Chemo-Immunotherapy
TME is a key determinant of ICI responsiveness, and dynamically changes alongside tumor progression. A pronounced synergistic effect between immunotherapy and chemotherapy may be achieved in mouse models wherein the immune system of the mice is intact. Nevertheless, current ICIs are usually administered to patients with advanced cancer, who exhibit a deteriorated immune system due to immunoediting and chemotherapy treatment. Theoretically, immunotherapy administered to patients in earlier stages of the disease, with less deteriorated immunity and before a myeloablative chemotherapy treatment, would be more likely to cause a durable immunity than that caused by most current regimens [61]. Likewise, first-line durvalumab in combination with etoposide plus platinum in treatment-naïve early-stage small-cell lung cancer (SCLC) showed an improvement in overall survival (OS) compared with chemotherapy alone [62]. A recent meta-analysis based on 12 phase-III clinical trials with 9236 metastatic NSCLC patients reported that the addition of chemotherapy to ICIs enhanced their treatment efficacy as a first-line treatment [63]. Nevertheless, this approach could have the disadvantage of exposing patients who would have responded to monotherapy to unnecessary toxicity.5.3. The Sequence of Chemo-Immunotherapy
Chemotherapy and immunotherapy are administered concurrently in the vast majority of clinical trials. Still, the sequence of their administration may meaningfully affect outcomes [64]. For instance, ipilimumab (anti-CTLA-4 antibody) administered after carboplatin and paclitaxel (but not concurrent administration) is associated with improved immune-related progression-free survival (PFS) in SCLC compared with chemotherapy alone [65]. By contrast, patients with metastatic melanoma who progress after PD-1 therapy benefit from the subsequent addition of chemotherapy [66]. Therefore, a rational timing selection is susceptible to becoming a cornerstone of chemo-immunotherapy success; intuitively, immunotherapy is more likely to work when administered before myeloablative chemotherapy regimens. Contrarily, non-myeloablative chemotherapy using drugs with immune stimulatory properties (i.e., causing ICD or a metronomic dose) are more likely to work before immunotherapy. Of note, doxorubicin and oxaliplatin, which are particularly efficient in promoting immune responses, are promising partners for administration before chemotherapy [67]. Enhancing lymphocyte recovery using immunomodulatory drugs or cytokines or minimizing chemotherapy-induced damage to the immune system may potentiate ICIs, and may be an alternative to a maximum tolerated dose of chemotherapy currently used in clinical practice. Unfortunately, few clinical trials have tried to systematically identify the optimal conditions for chemo-immunotherapy, and no consensus has yet been achieved regarding the right dose, timing, and sequence of chemo-immunotherapy combinations that may maximize their clinical benefits.References
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16.
- Pol, J.; Vacchelli, E.; Aranda, F.; Castoldi, F.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Fucikova, J.; Galon, J.; Spisek, R.; et al. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015, 4, e1008866.
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 924–937.
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156.
- Liu, P.; Chen, J.; Zhao, L.; Hollebecque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. PD-1 blockade synergizes with oxaliplatin-based, but not cisplatin-based, chemotherapy of gastric cancer. Oncoimmunology 2022, 11, 2093518.
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500.
- Zitvogel, L.; Pitt, J.M.; Daillere, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773.
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741.
- Limagne, E.; Thibaudin, M.; Nuttin, L.; Spill, A.; Derangere, V.; Fumet, J.D.; Amellal, N.; Peranzoni, E.; Cattan, V.; Ghiringhelli, F. Trifluridine/Tipiracil plus Oxaliplatin Improves PD-1 Blockade in Colorectal Cancer by Inducing Immunogenic Cell Death and Depleting Macrophages. Cancer Immunol. Res. 2019, 7, 1958–1969.
- Li, Y.; Zhang, H.; Li, Q.; Zou, P.; Huang, X.; Wu, C.; Tan, L. CDK12/13 inhibition induces immunogenic cell death and enhances anti-PD-1 anticancer activity in breast cancer. Cancer Lett. 2020, 495, 12–21.
- Fukushima, H.; Yoshida, S.; Kijima, T.; Nakamura, Y.; Fukuda, S.; Uehara, S.; Yasuda, Y.; Tanaka, H.; Yokoyama, M.; Matsuoka, Y.; et al. Combination of Cisplatin and Irradiation Induces Immunogenic Cell Death and Potentiates Postirradiation Anti-PD-1 Treatment Efficacy in Urothelial Carcinoma. Int. J. Mol. Sci. 2021, 22, 535.
- Shan, C.K.; Du, Y.B.; Zhai, X.T.; Wang, Y.X.; Li, Y.; Gong, J.H.; Ge, Z.J.; Liu, X.J.; Zhen, Y.S. Pingyangmycin enhances the antitumor efficacy of anti-PD-1 therapy associated with tumor-infiltrating CD8(+) T cell augmentation. Cancer Chemother. Pharmacol. 2021, 87, 425–436.
- Yamazaki, T.; Buque, A.; Ames, T.D.; Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology 2020, 9, 1721810.
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40.
- Janjigian, Y.Y.; Kawazoe, A.; Yanez, P.; Li, N.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.; Shen, L.; Tang, Y.; et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 2021, 600, 727–730.
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928.
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045.
- Demaria, S.; Volm, M.D.; Shapiro, R.L.; Yee, H.T.; Oratz, R.; Formenti, S.C.; Muggia, F.; Symmans, W.F. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin. Cancer Res. 2001, 7, 3025–3030.
- Muliaditan, T.; Opzoomer, J.W.; Caron, J.; Okesola, M.; Kosti, P.; Lall, S.; Van Hemelrijck, M.; Dazzi, F.; Tutt, A.; Grigoriadis, A.; et al. Repurposing Tin Mesoporphyrin as an Immune Checkpoint Inhibitor Shows Therapeutic Efficacy in Preclinical Models of Cancer. Clin. Cancer Res. 2018, 24, 1617–1628.
- Hong, M.; Puaux, A.L.; Huang, C.; Loumagne, L.; Tow, C.; Mackay, C.; Kato, M.; Prevost-Blondel, A.; Avril, M.F.; Nardin, A.; et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 2011, 71, 6997–7009.
- Kaneno, R.; Shurin, G.V.; Tourkova, I.L.; Shurin, M.R. Chemomodulation of human dendritic cell function by antineoplastic agents in low noncytotoxic concentrations. J. Transl. Med. 2009, 7, 58.
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309.
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br. J. Cancer 2010, 102, 115–123.
- Lacour, S.; Hammann, A.; Wotawa, A.; Corcos, L.; Solary, E.; Dimanche-Boitrel, M.T. Anticancer agents sensitize tumor cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated caspase-8 activation and apoptosis. Cancer Res. 2001, 61, 1645–1651.
- Ramakrishnan, R.; Assudani, D.; Nagaraj, S.; Hunter, T.; Cho, H.I.; Antonia, S.; Altiok, S.; Celis, E.; Gabrilovich, D.I. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J. Clin. Investig. 2010, 120, 1111–1124.
- Scurr, M.; Pembroke, T.; Bloom, A.; Roberts, D.; Thomson, A.; Smart, K.; Bridgeman, H.; Adams, R.; Brewster, A.; Jones, R.; et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 6771–6780.
- Dimeloe, S.; Frick, C.; Fischer, M.; Gubser, P.M.; Razik, L.; Bantug, G.R.; Ravon, M.; Langenkamp, A.; Hess, C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur. J. Immunol. 2014, 44, 3614–3620.
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282.
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118.
- Kanterman, J.; Sade-Feldman, M.; Biton, M.; Ish-Shalom, E.; Lasry, A.; Goldshtein, A.; Hubert, A.; Baniyash, M. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014, 74, 6022–6035.
- Huang, X.; Cui, S.; Shu, Y. Cisplatin selectively downregulated the frequency and immunoinhibitory function of myeloid-derived suppressor cells in a murine B16 melanoma model. Immunol. Res. 2016, 64, 160–170.
- Sevko, A.; Michels, T.; Vrohlings, M.; Umansky, L.; Beckhove, P.; Kato, M.; Shurin, G.V.; Shurin, M.R.; Umansky, V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471.
- Kwong, T.T.; Wong, C.H.; Zhou, J.Y.; Cheng, A.S.L.; Sung, J.J.Y.; Chan, A.W.H.; Chan, S.L. Chemotherapy-induced recruitment of myeloid-derived suppressor cells abrogates efficacy of immune checkpoint blockade. JHEP Rep. 2021, 3, 100224.
- Ding, Z.C.; Munn, D.H.; Zhou, G. Chemotherapy-induced myeloid suppressor cells and antitumor immunity: The Janus face of chemotherapy in immunomodulation. Oncoimmunology 2014, 3, e954471.
- Ding, Z.C.; Lu, X.; Yu, M.; Lemos, H.; Huang, L.; Chandler, P.; Liu, K.; Walters, M.; Krasinski, A.; Mack, M.; et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014, 74, 3441–3453.
- Wesolowski, R.; Duggan, M.C.; Stiff, A.; Markowitz, J.; Trikha, P.; Levine, K.M.; Schoenfield, L.; Abdel-Rasoul, M.; Layman, R.; Ramaswamy, B.; et al. Circulating myeloid-derived suppressor cells increase in patients undergoing neo-adjuvant chemotherapy for breast cancer. Cancer Immunol. Immunother. 2017, 66, 1437–1447.
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154.
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190.
- Acebes-Huerta, A.; Lorenzo-Herrero, S.; Folgueras, A.R.; Huergo-Zapico, L.; Lopez-Larrea, C.; Lopez-Soto, A.; Gonzalez, S. Drug-induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM-1 receptors. Oncoimmunology 2016, 5, e1074378.
- Zhang, X.; Wang, D.; Li, Z.; Jiao, D.; Jin, L.; Cong, J.; Zheng, X.; Xu, L. Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front. Immunol. 2020, 11, 331.
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648.
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3.
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153.
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668.
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319.
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vely, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbe, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977.
- Dong, W.; Wu, X.; Ma, S.; Wang, Y.; Nalin, A.P.; Zhu, Z.; Zhang, J.; Benson, D.M.; He, K.; Caligiuri, M.A.; et al. The Mechanism of Anti-PD-L1 Antibody Efficacy against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov. 2019, 9, 1422–1437.
- Pasquier, E.; Kavallaris, M.; Andre, N. Metronomic chemotherapy: New rationale for new directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465.
- Quartino, A.L.; Friberg, L.E.; Karlsson, M.O. A simultaneous analysis of the time-course of leukocytes and neutrophils following docetaxel administration using a semi-mechanistic myelosuppression model. Investig. New Drugs 2012, 30, 833–845.
- Tanaka, H.; Matsushima, H.; Mizumoto, N.; Takashima, A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009, 69, 6978–6986.
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol. 2019, 21, 730–741.
- Wu, J.; Jordan, M.; Waxman, D.J. Metronomic cyclophosphamide activation of anti-tumor immunity: Tumor model, mouse host, and drug schedule dependence of gene responses and their upstream regulators. BMC Cancer 2016, 16, 623.
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.J.; Spielbauer, K.; Van Waes, C.; Schmitt, N.C. Cisplatin Alters Antitumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151.
- Chen, Q.; Xia, R.; Zheng, W.; Zhang, L.; Li, P.; Sun, X.; Shi, J. Metronomic paclitaxel improves the efficacy of PD-1 monoclonal antibodies in breast cancer by transforming the tumor immune microenvironment. Am. J. Transl. Res. 2020, 12, 519–530.
- Skavatsou, E.; Semitekolou, M.; Morianos, I.; Karampelas, T.; Lougiakis, N.; Xanthou, G.; Tamvakopoulos, C. Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC. Cancers 2021, 13, 1901.
- Maharjan, R.; Choi, J.U.; Kweon, S.; Pangeni, R.; Lee, N.K.; Park, S.J.; Chang, K.Y.; Park, J.W.; Byun, Y. A novel oral metronomic chemotherapy provokes tumor specific immunity resulting in colon cancer eradication in combination with anti-PD-1 therapy. Biomaterials 2022, 281, 121334.
- Petrizzo, A.; Mauriello, A.; Luciano, A.; Rea, D.; Barbieri, A.; Arra, C.; Maiolino, P.; Tornesello, M.; Gigantino, V.; Botti, G.; et al. Inhibition of tumor growth by cancer vaccine combined with metronomic chemotherapy and anti-PD-1 in a pre-clinical setting. Oncotarget 2018, 9, 3576–3589.
- Katsumata, N.; Yasuda, M.; Takahashi, F.; Isonishi, S.; Jobo, T.; Aoki, D.; Tsuda, H.; Sugiyama, T.; Kodama, S.; Kimura, E.; et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: A phase 3, open-label, randomised controlled trial. Lancet 2009, 374, 1331–1338.
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, eaax0182.
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Ozguroglu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939.
- Dafni, U.; Tsourti, Z.; Vervita, K.; Peters, S. Immune checkpoint inhibitors, alone or in combination with chemotherapy, as first-line treatment for advanced non-small cell lung cancer. A systematic review and network meta-analysis. Lung Cancer 2019, 134, 127–140.
- Kwon, M.; Jung, H.; Nam, G.H.; Kim, I.S. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J. Control. Release 2021, 331, 321–334.
- Reck, M.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Lu, H.; Cuillerot, J.M.; Lynch, T.J. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase 2 trial. Ann. Oncol. 2013, 24, 75–83.
- Yan, Y.; Cao, S.; Liu, X.; Harrington, S.M.; Bindeman, W.E.; Adjei, A.A.; Jang, J.S.; Jen, J.; Li, Y.; Chanana, P.; et al. CX3CR1 identifies PD-1 therapy-responsive CD8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight 2018, 3, e97828.
- Galluzzi, L.; Humeau, J.; Buque, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741.
More