Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Pietro Desimone and Version 2 by Camila Xu.
Arrhythmogenic substrate, modulating factors, and triggering factors (the so-called Coumel’s triangle concept) play a primary role in atrial fibrillation (AF) pathophysiology. Several years have elapsed since the concept of the relevance of autonomic nervous system (ANS) influences on atrial cells’ electrophysiological characteristics was advanced.
- atrial fibrillation
- autonomic nervous system
- sympathetic
- parasympathetic
1. Introduction
Arrhythmogenic substrate, modulating factors, and triggering factors (the so-called Coumel’s triangle concept) play a primary role in atrial fibrillation (AF) pathophysiology. Several years have elapsed since Coumel et al. [1] advanced the concept of the relevance of autonomic nervous system (ANS) influences on atrial cells’ electrophysiological characteristics. The ANS is not only associated with cardiac rhythm regulation but also exerts an important role in the triggering and maintenance of atrial fibrillation. The heart is richly innervated by the autonomic system that can be located either inside the heart (intrinsic ANS) or in brainstem and cardiac preganglionic fibers (extrinsic ANS). Both intrinsic and extrinsic autonomic nervous system divisions are fundamental for cardiac function and arrhythmogenesis [2][3][4][2,3,4].
2. The Biomolecular Explanation of the Autonomic Nervous System Role in Coumel’s Triangle
Each of the myocardial sleeves of the pulmonary veins (PVs) is richly innervated by four of the left atrial GP [5]. These four GPs are localized in areas of fractioned atrial potentials, as widely described in the literature [6]. Po et al. demonstrated that the injection of acetylcholine into the GP can induce focal firing coming from adjacent pulmonary veins [7]. Moreover, in animal models of PV preparations with a surrounding left atrial myocardial tissue, local autonomic stimulation induces early afterdepolarizations (EADs) and bursts of triggered firing from the PVs [8]. This triggered activation was similar to the pattern recorded from the PVs in patients with paroxysmal AF [6]. It must be underlined that in autonomic nerve structures, there is a frequent co-localization of sympathetic and parasympathetic fibers making a complete selective radiofrequency catheter ablation extremely difficult. The spatial and functional interplay between sympathetic and parasympathetic fibers plays a relevant role in starting and maintaining the AF burden [9]; for example, sympathetic stimulation would promote cholinergic-mediated AF initiation [10].2.1. Cardiac Autonomic Neurotransmission: Adrenergic and Cholinergic Molecular Pathways
The effect of the autonomic nervous system on cardiomyocytes is mainly related to changes in ion channel functioning, leading to action potential length, conductive forces, and conduction velocity modifications (Figure 1). The extremely complex mechanism of neurotransmitter production, release, and reuptake is highly regulated [11].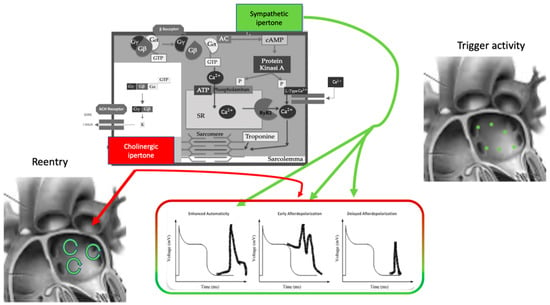
Figure 1. Autonomic nervous system and atrial fibrillation. Biomolecular mechanisms and the influence of the ANS on triggers and atrial reentry mechanisms. See text for explanation.
2.2. Interplay between Autonomic Nervous System and Cardiomyocytes Action Potential
The autonomic nervous system promotes AF by focal or reentrant mechanisms (Figure 1) [17][18][17,18]. Adrenergic activation may promote focal activity by three main principal cellular mechanisms: enhanced automaticity, early afterdepolarization (EAD), or delayed afterdepolarization-associated triggered activity (DAD) [19]. Adrenergic activation causes increased automaticity through the prevention of spontaneous phase 4 depolarization which is promoted by If and inhibited by the IK1 current. Moreover, beta-adrenergic activation may raise focal activity through two principal cellular mechanisms: early afterdepolarization or delayed afterdepolarization-associated triggered activity [20]. Norepinephrine can lead to EAD in a setting of prolonged or shortened action potential duration (APD). β-adrenergic activation through PKA/CaMKII phosphorylation enhances plateau ICa, leading to an increased phase 2 EAD likelihood, especially in individuals suffering from long-QT syndrome, in whom adrenergic augmentation of IKs is deficient [21]. Phase3-EAD is associated with increased Ca2+ transient and shortened APD: it is caused by the simultaneous activation of the sympathetic nervous system with increased Ca2+ and parasympathetic nervous system activation of IKAch. The simultaneous presence of short APD and increased Ca2+ transient leads to late-phase 3 EAD and trigger arrhythmias [8][22][23][8,22,23]. As a matter of fact, APD prolongation permits L-type Ca2+ channels to recover from inactivation, leading to an inward current, causing an EAD. B-adrenergic stimulation alone can also lead to phase 2 EAD in a setting of prolonged APD by increasing the depolarizing currents (L-type Ca2+ current [ICa, L]) [19]. DAD results from the dysfunction of channel type 2 ryanodine receptor (RyR2) caused by the increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II (CaMKII); it promotes SR Ca2+ release and Na+/Ca2+ exchanger (NCX) activation. All these processes produce a depolarizing transient inward current (Iti) that causes delayed afterdepolarization. The molecular basis of maintaining reentry mechanisms is not clearly understood; however, in all conceptual models, functional reentry is promoted by atrial refractoriness [24]. Vagal stimulation abbreviates atrial refractoriness by increasing IKACh. In addition, atrial refractoriness abbreviation made by parasympathetic stimulation is characterized by a relevant regional variability that perpetuates the vagal AF-promoting effects. The role of increased refractoriness spatial heterogeneity is confirmed in animal studies in which flecainide terminated the vagus-related pro-arrhythmic effects on AF [25]. A very interesting observation was that, in an ion channel model, a reentry spiral wave induced by a sympathetic stimulation tended to organize and terminate immediately, whereas that induced by a vagal stimulation tended to be maintained in the presence of the refractoriness spatial dispersion. Therefore, this is the explanation as to why an adrenergic AF usually terminates spontaneously, while a vagal AF tends to be maintained [26]. Finally, Figure 2 illustrates a proposed point of view regarding the systemic role of the ANS in Coumel’s triangle. The hypothesis of an “Autonomic Coumel Triangle” stems from the condition of the fundamental role of ANS in all phases of the pathophysiological mechanism of AF. An increase in sympathetic tone favors trigger activity (e.g., ectopic activity from the pulmonary veins, but also from the right atrium) through three known mechanisms: enhanced automatism, EAD, and DAD. Together with sympathetic activation, vagal activity can contribute to triggering activity through the creation of a late-phase 3 EAD. This mechanism is very often present in paroxysmal AF in both healthy and young hearts. However, the role of ANS is also fundamental in forms of persistent AF and/or standing persistent AF where the substrate prevails. In these patients, the role of parasympathetic activity in favoring a spatially heterogeneous action potential and refractory period abbreviation that promotes the occurrence and maintenance of reentrant activity is of crucial importance. At the same time, sympathetic hyperactivity is able to contribute to arrhythmic substrate by promoting increased Ca2+/calmodulin binding and oxidative stress.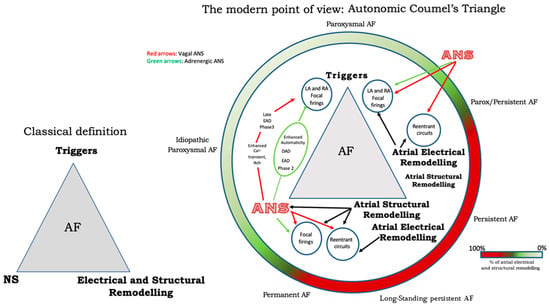
Figure 2.
The Autonomic Coumel’s Triangle. See text for explanation.