2. Cardiac Adaptations
2.1. Exercise Preserves Cardiomyocyte Ultrastructure
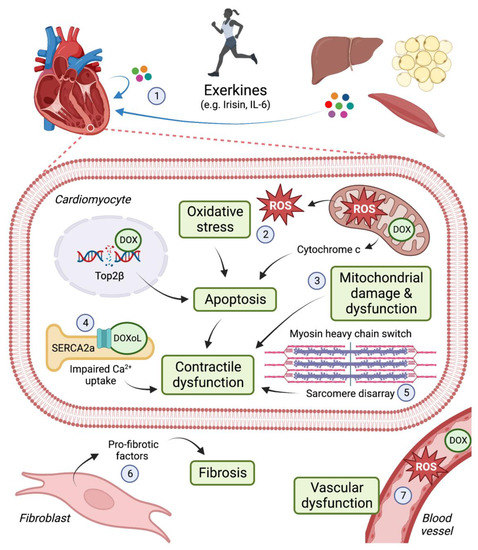
The cardiomyocyte cell population is responsible for heart contractility. Cardiomyocytes are composed of myofibrils that run parallel to each other, forming muscle fibres. Myofibrils are composed of repeating contractile units termed sarcomeres, defined by two consecutive Z lines. During cardiomyocyte contraction, the distance between the Z lines shorten. This is due to myosin and actin filaments sliding within the sarcomere. Specifically, actin filaments slide along myosin filaments as a result of actin-myosin cross-bridge cycling. The M line running through the middle of the sarcomere, composed of myomesin, and the Z lines on either side, function to hold the actin and myosin filaments in place. Impairment of sarcomeric structure and function is associated with diminished cardiac contraction (systolic dysfunction) and/or chamber filling (diastolic dysfunction).
Sequeira et al., examined the effects of 10 mg/kg of DOX (cumulative) on the ventricular cardiomyocyte ultrastructure in female rats
[24][39]. Cardiomyocytes from DOX-treated animals had disorganised myofibrils and sarcomeres, fragmented actin and myosin filaments and wavy Z lines with some areas showing an absence of Z lines. In contrast, rats that performed forced treadmill running throughout the course of the DOX treatment showed preserved myofibril integrity and improved sarcomeric organisation with reduced fragmentation of myofilaments. The data from this study suggest that aerobic exercise performed concurrent with DOX therapy is able to preserve the ultrastructural integrity of cardiomyocytes and ultimately preserve cardiomyocyte contractility.
2.2. Exercise Prevents MHC Isoform Shifts
Alterations in the composition of sarcomeres may contribute to DOX-induced reductions in contractility. In humans, α-myosin heavy chain (MHC) is the predominant MHC isoform in atria, while β-MHC is the predominant isoform in the ventricles
[25][91]. Smaller mammals, such as rodents, exhibit higher cardiac energetics and predominantly express the α-MHC isoform in the ventricles. Increasing the ratio of β-MHC to α-MHC reduces overall energy requirements but is associated with reduced contractility.
TIn this context, the peak power output of cardiomyocytes exclusively expressing the β-MHC isoform was ~50% lower than cardiomyocytes expressing 12% α-MHC and 88% β-MHC
[26][92].
It has previously been shown that DOX treatment upregulates β-MHC expression in the left ventricle of male rodents
[27][28][29][30][93,94,95,96]. Exercise preconditioning for 10 weeks significantly attenuated the expression of β-MHC in male rats administered a DOX dose of 10 mg/kg
[27][93]. The same group showed that DOX administered daily (1 mg/kg) for a period of 10 days increased β-MHC expression in the left ventricle. This response to DOX treatment was somewhat blunted in animals that underwent either forced or voluntary wheel running for 10 weeks prior to DOX
[28][94]. Similar findings were reported in a rat model of resistance training
[31][32][97,98]. Thus, exercise training may preserve cardiac contractility in part by maintaining a normal α-MHC/β-MHC ratio.
2.3. Exercise Alleviates Fibrosis
Cardiac fibrosis is a term used to describe the pathological process of scar tissue formation and interstitial collagen deposition that can lead to myocardial stiffness and dysfunction. The extracellular matrix (ECM) describes the network of non-cellular fibrous and non-fibrous proteins contributing to the maintenance of tissue architecture. Collagen is the most abundant fibrous ECM protein, accounting up to 30% of total protein found in the human body
[30][96]. The upregulation of collagen as a means of tissue repair can result in collagen accumulation and fibrosis formation. Clinically, anthracycline-based therapy has shown to increase the cardiac extracellular volume fraction (marker of fibrosis) as early as three months from treatment initiation
[31][32][97,98]. Similarly, histological analyses of rodent hearts show extensive fibrosis induced by DOX therapy
[24][33][34][35][39,59,99,100].
The anti-fibrotic effects of exercise performed concurrent with DOX therapy has been evaluated in four studies. Sequeira et al., reported significant myocardial fibrosis in female rats that received a DOX dose of 10 mg/kg (cumulative)
[24][39]. Moderate aerobic exercise training prevented this DOX-induced increase in fibrosis, evident by preserved connective tissue volume density. Similarly, Wang et al., observed significant collagen deposition in female mice immediately after DOX therapy, which persisted for 12 weeks
[33][59]. The authors did not observe any significant fibrosis in animals that underwent treadmill walking during DOX therapy, demonstrating that exercise can prevent the development of fibrosis in this setting. Interestingly, low-to-moderate aerobic exercise training was unable to prevent myocardial fibrosis or preserve LVEF in male mice that received a DOX dose of 25 mg/kg (cumulative)
[34][99]. Yang et al., investigated signalling pathways contributing to DOX-induced fibrosis in male rats
[35][100]. DOX induced the expression of fibrotic markers, namely transforming growth factor beta 1 (TGF-β1), connective tissue growth factor (CTGF), phosphorylated extracellular signal-regulated kinase (p-ERK) and specific protein 1 (Sp1). Moderate intensity aerobic exercise during DOX therapy was able to mitigate the expression of these fibrotic factors, thereby reducing fibrosis and preserving cardiac function.
2.4. Exercise Preserves Cardiac Size
Cardiac atrophy, or a reduction in heart mass and size, has been reported in both children and adults undergoing DOX therapy
[36][37][101,102]. In survivors of childhood cancer, DOX-associated cardiotoxicity can manifest as restrictive cardiomyopathy, evident 15 years post-exposure
[36][101]. This condition is characterised by increased myocardial stiffness and reduced left ventricular dimensions. Among anthracycline-treated adults who develop cardiomyopathy, a dose-dependent reduction in left ventricular mass is observed
[38][39][103,104]. The extent to which this is due to decreased cardiomyocyte size, as opposed to increased cardiomyocyte death, is not fully understood.
DOX contributes to cardiac atrophy by disrupting the balance of protein synthesis and degradation
[38][39][103,104]. The degradation of proteins is regulated by the ubiquitin proteasome system (UPS) and the autophagic-lysosomal pathway (ALP
[40][105]. Ubiquitin-labelled proteins are degraded by the 26S proteosome, while ALP-mediated protein degradation involves the sequestration of proteins within lysosomes via microautophagy, macroautophagy or chaperone-mediated autophagy
[40][105]. UPS-mediated protein degradation was observed in neonatal rat ventricular myocytes treated with a clinically-relevant concentration of DOX
[38][103]. This may be a mechanism by which DOX contributes to cardiomyocyte atrophy, although cell size was not reported in the above study. DOX also stimulates autophagy in both cultured cardiomyocytes and in mouse hearts; however, impairments in lysosomal function block autophagic flux, leading to the accumulation of autolysosomes, ROS production and contractile dysfunction
[39][104]. Four weeks of DOX treatment (10 mg/kg cumulative dose) increased the formation of autophagosomes in hearts of young female mice, an effect that was completely abolished with a 2-week aerobic exercise intervention
[33][59]. The mechanisms by which exercise preserves autophagic signalling in settings of DOX-induced cardiotoxicity have not been extensively investigated, but exercise is known to alter autophagic gene expression in the heart
[41][106] and 5 days of treadmill running prior to an acute DOX challenge (20 mg/kg, male rats) prevented DOX-induced increases in the expression of autophagy proteins (Beclin 1, ATG4, ATG7, ATG12 and LC3) and the lysosomal proteases cathepsin B and cathepsin L
[42][107].
DOX-induced cardiotoxicity is characterised by a decrease in cardiomyocyte area and volume, which reduces the overall heart weight/body weight (HW/BW) ratio
[24][34][43][39,99,108]. HW/BW ratio was 34% lower in male rats receiving DOX (4 mg/kg/week for 4 weeks) compared with controls, an effect that was prevented by exercise preconditioning
[43][108]. Gomez-Santos et al., demonstrated significant reductions in left ventricular mass and cardiomyocyte area with 5 weeks of DOX treatment (5 mg/kg/week) in male mice
[34][99]. Low-to-moderate aerobic exercise performed concurrent with DOX treatment blunted cardiac and cardiomyocyte atrophy in this model. Similarly, aerobic exercise concurrent with DOX treatment prevented the decline in cardiomyocyte volume density observed in female rats
[24][39]. These studies demonstrate that exercise performed prior to or during DOX therapy can preserve cardiomyocyte size and prevent cardiac atrophy.
2.5. Exercise Preserves SERCA2A Activity
Impairment of calcium homeostasis has been implicated in the pathophysiology of DOX-associated cardiotoxicity
[44][109]. Regulation of calcium homeostasis is controlled by the sarcoplasmic reticulum (SR), a specialised endoplasmic reticulum that surrounds the contractile myofilaments of muscle cells. The SR ATPase 2a (SERCA2a) calcium pump, expressed in cardiomyocytes and type I skeletal muscle, is a key protein that pumps calcium back into the SR, leading to myocyte relaxation. Thus, reduced activity or expression of SERCA2a contributes to impaired calcium handling, relaxation and contractility, while interventions that increase SERCA2a expression improve cardiac function in animal models of HF
[45][110]. DOXoL, a metabolite of DOX, interacts directly with SERCA2a and is a potent inhibitor of SERCA2a activity and SR calcium uptake
[46][111]. Interestingly, inhibition of calcium uptake by DOXoL was almost 100 times greater than that of the parent compound (DOX).
The role of exercise in regulating SERCA2a expression in settings of DOX-induced cardiotoxicity has previously been investigated. Hydock et al., observed an ~80–90% decrease in SERCA2a expression in male rats receiving a daily DOX injection of 1 mg/kg for a period of 10 days
[28][94]. Long-term exercise preconditioning (10 weeks forced or voluntary wheel running) protected rats from developing diastolic dysfunction in response to DOX exposure; however, this occurred independently of an increase in SERCA2a levels, indicating that other mechanisms may be contributing to the preservation of cardiac function in this setting. Lien et al., reported significant reductions in SERCA2a expression in male rats administered a single injection of DOX at a dose of 10 or 15 mg/kg
[20]. Forced treadmill running or voluntary wheel running for 5 days prior to DOX injection attenuated DOX-induced reductions in SERCA2a expression, with treadmill running providing the most benefit. This was associated with improvements in diastolic and systolic function, for both exercise modalities. Dolinsky et al., reported that exercise training concurrent with DOX treatment (8 mg/kg/week) partially prevented DOX-induced reductions in SERCA2a expression in female mice
[47][38]. Together, these studies reveal that exercise can preserve SERCA2a expression levels and thus attenuate DOX-induced cardiac dysfunction.