Parkinson’s disease (PD) is a multifactorial disorder involving both motor and non-motor symptoms caused by the progressive death of distinct neuronal populations, including dopaminergic neurons in the substantia nigra. The deposition of aggregated α-synuclein protein into Lewy body inclusions is a hallmark of the disorder, and α-synuclein pathology has been found in the enteric nervous system (ENS) of PD patients up to two decades prior to diagnosis. In combination with the high occurrence of gastrointestinal dysfunction in early stages of PD, evidence strongly suggests that some forms of PD may originate in the gut.
- alpha-synuclein
- Parkinson’s disease
- enteric nervous system
1. Introduction
2. The Human Enteric Nervous System
The ENS is an intricate network of neuronal cell bodies and fibers that perform a wide array of digestive functions, including moving food through the gastrointestinal tract, facilitating nutrient uptake, regulating local blood flow, and supporting the immune system [38]. While the ENS is able to function independently of the CNS, bidirectional communication between the ENS and CNS serves to relay important information that ultimately affects organismal behavior and gastrointestinal functioning. The gut-brain axis involves the delivery of sensory information to the brain via spinal and vagal afferent pathways, and efferent motor signals to the gut by way of sympathetic and parasympathetic divisions of the autonomic nervous system [39]. Parasympathetic innervation via the vagal nerve originates in preganglionic neurons of the DMN, which synapse onto postganglionic neurons of the ENS. The nucleus ambiguus in the brainstem also supplies vagal motor efferents specifically to the pharynx and esophagus [40]. Vagal innervation of the ENS is densest in the upper gastrointestinal tract at the level of the esophagus and stomach and decreases more distally. With little to no vagal input to the distal colon and rectum, these regions are primarily regulated by the sacral parasympathetic nucleus of the spinal cord. The activity of the DMN and nucleus ambiguus can be modulated by sensory information that is relayed from the ENS through vagal afferent pathways to the nucleus of the solitary tract in the brainstem [40]. While the influence of parasympathetic pathways can result in both enhancement and suppression of gut motility, sympathetic innervation of the digestive tract mainly acts to inhibit motility as a pro-survival reflex that is mediated by prevertebral sympathetic ganglia [41]. Within the ENS, two main neuronal networks perform the complex integration of all local (intrinsic) neuronal activity, input from extrinsic sympathetic and parasympathetic neurons, as well as cues from the gastrointestinal environment. These networks, known as the myenteric (or Auerbach’s) plexus, and the submucosal (or Meissner’s) plexus, use the integrated input to determine their own sensory, motor, and secretory output. The ENS is located in the gastrointestinal wall, which is comprised of four main layers: the mucosa, submucosa, muscular layer, and adventitia or serosa (Figure 1). The layer closest to the lumen of the gut, the mucosa, can be further divided into the epithelium, which forms the lining of the mucosa; the lamina propria, which is made up of connective tissue; and the muscularis mucosae, a layer of smooth muscle.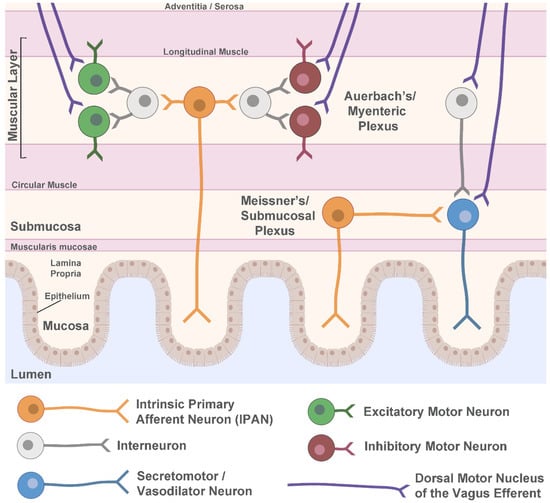
3. Lewy Pathology in the Enteric Nervous System in PD
4. Does α-Synuclein Aggregation Begin in the Gut and Spread to the Brain?
4.1. Evidence from Incidental LB Disease and Prodromal PD
4.2. Evidence from Human Vagotomy Studies
Besides the presence of Lewy pathology in the ENS, another key aspect of the gut-to-brain hypothesis of PD is the transmission of α-synuclein through the vagal nerve. The higher density of ENS pathology in the esophagus and stomach compared to lower regions in PD [11][12][13][16][11,12,13,16] is consistent with the high concentration of vagal inputs to these areas. PD patient vagotomy studies may, therefore, shed light on the dependence of the disease on intact vagal innervation of the gut. A former treatment for peptic ulcer, vagotomy, is the severing of the vagal nerve either fully (as in truncal vagotomy), solely to the stomach (selective vagotomy), or most selectively to the fundus and body of the stomach only (superselective vagotomy). While only a few studies[50][51][52][53] [50,51,52,53] have examined the potential relationship of vagal denervation with PD, the findings generally support a protective effect, potentially by reducing the ability of α-synuclein to invade the CNS.4.3. Prion-Like Transmission of α-Synuclein in Rodents and Monkeys
There is now extensive evidence from animal models supporting the theory of prion-like transmission of α-synuclein in PD and other synucleinopathies. In mice and rats, intracerebral inoculation of recombinant α-synuclein pre-formed fibrils (PFFs), brain tissue from symptomatic α-synuclein transgenic mice, or brain tissue from human synucleinopathy patients, results in widespread deposition of LB-like inclusions that are often associated with neurodegeneration and motor dysfunction [46][54][55][56][57][58][59][60][54,55,56,57,58,59,46,60]. Moreover, it appears that, regardless of the site of injection, aggregation propagates along synaptic connections and requires the presence of endogenous α-synuclein, similar to prions [54][55][57][58][59][60][54,55,57,58,59,60]. In macaque monkeys, injection of PD brain tissue containing insoluble LBs into the substantia nigra or striatum caused a loss of striatal terminals followed by dopamine neuron death and diffuse α-synuclein deposits in the remaining nigral cells [55].4.4. C. elegans as a Powerful Model System to Study α-Synuclein Pathogenicity in PD
While rodent and non-human primate models provide essential information with regards to how α-synuclein can behave in a mammalian system, complementary animal models that offer a rapidly aging nervous system and high genetic tractability are necessary to accelerate the discovery of disease mechanisms and potential treatments. The small nematode worm, C. elegans, provides such a platform, having a well-defined nervous system that gives rise to a complex set of behaviors [61], orthologs for 60–80% of human genes [62], conserved neurotransmitter signaling [63], and suitability to rapid large-scale behavioral and phenotypic screening approaches [61]. C. elegans is a premier model system to study aging and age-related disease, due to its short lifespan (2-4 weeks) and stereotyped age-dependent decline at the tissue, cellular, and molecular levels [64]. In addition, transgenic expression of human α-synuclein in worms has recapitulated progressive age-dependent neuron death, protein aggregation, and behavioral deficits [65][66][67][65,66,67], and proven useful for the study of cell autonomous disease mechanisms in dopaminergic neurons [68][69][70][68,69,70].In an effort to generate prion-like α-synuclein transmission models initiated in the gut of C. elegans, aour group recently published the neurotoxic effects of feeding worms human α-synuclein PFFs [71]. To our knowledge, this is the first report of α-synuclein PFF exposure in C. elegans. Similar to mouse models, scholarswe found that PFF ingestion in C. elegans promotes dopaminergic neurodegeneration, accelerates the aggregation of host α-synuclein in muscle, and induces an age-dependent motor decline. The development of these new models may serve to complement existing rodent model systems by acting as platforms for high-throughput discovery.
4.5. Alternative Hypotheses of α-Synuclein Spreading in PD
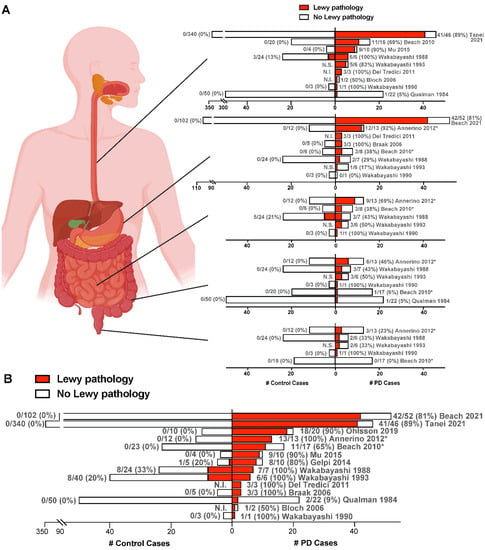
Despite mounting evidence in humans and animal models supporting the gut-to-brain hypothesis of α-synuclein transmission in PD, alternative possibilities have been proposed that fuel ongoing debate. A major criticism of the gut-origin hypothesis of PD is the lack of individuals found to have α-synuclein pathology in the ENS in the absence of pathology in the CNS. It would be expected that if α-synuclein pathology begins in the ENS and spreads to the CNS via the vagal nerve, there should be normal subjects with undiagnosed, prodromal PD that harbor ENS and/or vagal nerve pathology without evidence of lesions in the CNS. To address this issue, Beach and colleagues [43] conducted an autopsy study of stomach and/or vagal nerve tissue from 111 normal elderly controls that had no CNS pathology, 33 ILBD cases with some CNS pathology, and 53 confirmed PD cases. None of the normal subjects were found to have α-synuclein lesions in the stomach or vagal tissue, whereas 17% and 81% of ILBD and PD cases, respectively, had stomach pathology, and 46% and 89% of ILBD and PD cases, respectively, had vagal pathology. However, the low rate of prodromal PD estimated to exist in the aged population drastically reduces the probability of detection. Rather than arguing specifically against the gut-to-brain hypothesis of PD, the discovery of pathological α-synuclein in the vagus nerve of the majority of PD patients and almost half of ILBD patients can alternatively be interpreted as supporting the vagus nerve acting as a conduit for α-synuclein transmission between the gut and the brain, potentially in either direction.References
- Ding, C.; Wu, Y.; Chen, X.; Chen, Y.; Wu, Z.; Lin, Z.; Kang, D.; Fang, W.; Chen, F. Global, regional, and national burden and attributable risk factors of neurological disorders: The Global Burden of Disease study 1990–2019. Front. Public Health 2022, 10, 952161. Ding, C.; Wu, Y.; Chen, X.; Chen, Y.; Wu, Z.; Lin, Z.; Kang, D.; Fang, W.; Chen, F. Global, regional, and national burden and attributable risk factors of neurological disorders: The Global Burden of Disease study 1990–2019. Front. Public Health 2022, 10, 952161.
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442. Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442.
- Lim, S.Y.; Fox, S.H.; Lang, A.E. Overview of the extranigral aspects of Parkinson disease. Arch. Neurol. 2009, 66, 167–172. Lim, S.Y.; Fox, S.H.; Lang, A.E. Overview of the extranigral aspects of Parkinson disease. Arch. Neurol. 2009, 66, 167–172.
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nature reviews. Neuroscience 2013, 14, 626–636.
- Fasano, A.; Visanji, N.P.; Liu, L.W.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639.
- Bullich, C.; Keshavarzian, A.; Garssen, J.; Kraneveld, A.; Perez-Pardo, P. Gut Vibes in Parkinson’s Disease: The Microbiota-Gut-Brain Axis. Mov. Disord. Clin. Pract. 2019, 6, 639–651.
- Qualman, S.J.; Haupt, H.M.; Yang, P.; Hamilton, S.R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s disease. Gastroenterology 1984, 87, 848–856.
- Kupsky, W.J.; Grimes, M.M.; Sweeting, J.; Bertsch, R.; Cote, L.J. Parkinson’s disease and megacolon: Concentric hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology 1987, 37, 1253–1255.
- Wakabayashi, K.; Takahashi, H.; Takeda, S.; Ohama, E.; Ikuta, F. Parkinson’s disease: The presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 1988, 76, 217–221.
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Ikuta, F. Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990, 79, 581–583.
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Takeda, S.; Ikuta, F. Lewy bodies in the visceral autonomic nervous system in Parkinson’s disease. Adv. Neurol. 1993, 60, 609–612.
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72.
- Bloch, A.; Probst, A.; Bissig, H.; Adams, H.; Tolnay, M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol. Appl. Neurobiol. 2006, 32, 284–295.
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White, C.L., III; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Arizona Parkinson’s Disease Consortium Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702.
- Del Tredici, K.; Duda, J.E. Peripheral Lewy body pathology in Parkinson’s disease and incidental Lewy body disease: Four cases. J. Neurol. Sci. 2011, 310, 100–106.
- Annerino, D.M.; Arshad, S.; Taylor, G.M.; Adler, C.H.; Beach, T.G.; Greene, J.G. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol. 2012, 124, 665–680.
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1010–1018.
- Mu, L.; Chen, J.; Sobotka, S.; Nyirenda, T.; Benson, B.; Gupta, F.; Sanders, I.; Adler, C.H.; Caviness, J.N.; Shill, H.A.; et al. Arizona Parkinson’s Disease Consortium Alpha-Synuclein Pathology in Sensory Nerve Terminals of the Upper Aerodigestive Tract of Parkinson’s Disease Patients. Dysphagia 2015, 30, 404–417.
- Beach, T.G.; Corbillé, A.G.; Letournel, F.; Kordower, J.H.; Kremer, T.; Munoz, D.G.; Intorcia, A.; Hentz, J.; Adler, C.H.; Sue, L.I.; et al. Multicenter Assessment of Immunohistochemical Methods for Pathological Alpha-Synuclein in Sigmoid Colon of Autopsied Parkinson’s Disease and Control Subjects. J. Park. Dis. 2016, 6, 761–770.
- Ohlsson, B.; Englund, E. Atrophic Myenteric and Submucosal Neurons Are Observed in Parkinson’s Disease. Park. Dis. 2019, 2019, 7935820.
- Tanei, Z.I.; Saito, Y.; Ito, S.; Matsubara, T.; Motoda, A.; Yamazaki, M.; Sakashita, Y.; Kawakami, I.; Ikemura, M.; Tanaka, S.; et al. Lewy pathology of the esophagus correlates with the progression of Lewy body disease: A Japanese cohort study of autopsy cases. Acta Neuropathol. 2021, 141, 25–37.
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938.
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737.
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 39.
- Hasegawa, S.; Goto, S.; Tsuji, H.; Okuno, T.; Asahara, T.; Nomoto, K.; Shibata, A.; Fujisawa, Y.; Minato, T.; Okamoto, A.; et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS ONE 2015, 10, e0142164.
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 2016, 32, 66–72.
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 739–749.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108.
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173.
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064.
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. French Parkinson’s Disease Genetics Study Group G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471.
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5.
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Fleming, M.A., 2nd; Ehsan, L.; Moore, S.R.; Levin, D.E. The Enteric Nervous System and Its Emerging Role as a Therapeutic Target. Gastroenterol. Res. Pract. 2020, 2020, 8024171.
- Gershon, M.D.; Margolis, K.G. The gut, its microbiome, and the brain: Connections and communications. J. Clin. Investig. 2021, 131, e143768.
- Chang, H.Y.; Mashimo, H.; Goyal, R.K. Musings on the wanderer: What’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G357–G366.
- Benarroch, E.E. Enteric nervous system: Functional organization and neurologic implications. Neurology 2007, 69, 1953–1957.
- Mogor, F.; Kovács, T.; Lohinai, Z.; Dora, D. The Enteric Nervous System and the Microenvironment of the Gut: The Translational Aspects of the Microbiome-Gut-Brain Axis. Appl. Sci. 2021, 11, 12000.
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Shill, H.A.; Driver-Dunckley, E.; Mehta, S.H.; Intorcia, A.J.; Glass, M.J.; Walker, J.E.; Arce, R.; et al. Vagus Nerve and Stomach Synucleinopathy in Parkinson’s Disease, Incidental Lewy Body Disease, and Normal Elderly Subjects: Evidence Against the “Body-First” Hypothesis. J. Park. Dis. 2021, 11, 1833–1843.
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320.Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320.
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.C.; Citron, M.; Biere, A.L. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’ disease. J. Biol. Chem. 1999, 274, 19509–19512.Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.C.; Citron, M.; Biere, A.L. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’ disease. J. Biol. Chem. 1999, 274, 19509–19512.
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725.
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015.
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503.
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506.
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529.
- Tysnes, O.B.; Kenborg, L.; Herlofson, K.; Steding-Jessen, M.; Horn, A.; Olsen, J.H.; Reichmann, H. Does vagotomy reduce the risk of Parkinson’s disease? Ann. Neurol. 2015, 78, 1011–1012.
- Borghammer, P. How does Parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 48–57.
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002.
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. Peelaerts,W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344.
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362.
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560.
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain J. Neurol. 2013, 136 Pt 4, 1128–1138. Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain J. Neurol. 2013, 136 Pt 4, 1128–1138.
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986.
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953.
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199.
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent Window into Biology: A Primer on Caenorhabditis Elegans. WormBook: The Online Review of C. Elegans Biology. 2015, pp. 1–31. Available online: http://www.wormbook.org/chapters/www_celegansintro/celegansintro.html (accessed on 20 February 2023).Corsi, A.K.;Wightman, B.; Chalfie, M. A TransparentWindow into Biology: A Primer on Caenorhabditis elegans. WormBook: The Online Review of C. elegans Biology. 2015, pp. 1–31. Available online: http://www.wormbook.org/chapters/www_celegansintro/celegansintro.html (accessed on 20 February 2023).
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–398. Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–398.
- Hobert, O. The Neuronal Genome of Caenorhabditis Elegans. WormBook: The Online Review of C. Elegans Biology. 2013, pp. 1–106. Available online: http://www.wormbook.org/chapters/www_neuronalgenome/neuronalgenome.html (accessed on 20 February 2023). Hobert, O. The Neuronal Genome of Caenorhabditis elegans. WormBook: The Online Review of C. elegans Biology. 2013, pp. 1–106. Available online: http://www.wormbook.org/chapters/www_neuronalgenome/neuronalgenome.html (accessed on 20 February 2023).
- Son, H.G.; Altintas, O.; Kim, E.J.E.; Kwon, S.; Lee, S.V. Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell 2019, 18, e12853. Son, H.G.; Altintas, O.; Kim, E.J.E.; Kwon, S.; Lee, S.V. Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell 2019, 18, e12853.
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172. Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172.
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A.C. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027. Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027.
- Cao, P.; Yuan, Y.; Pehek, E.A.; Moise, A.R.; Huang, Y.; Palczewski, K.; Feng, Z. Alpha-synuclein disrupted dopamine homeostasis leads to dopaminergic neuron degeneration in Caenorhabditis elegans. PLoS ONE 2010, 5, e9312. Cao, P.; Yuan, Y.; Pehek, E.A.; Moise, A.R.; Huang, Y.; Palczewski, K.; Feng, Z. Alpha-synuclein disrupted dopamine homeostasis leads to dopaminergic neuron degeneration in Caenorhabditis elegans. PLoS ONE 2010, 5, e9312.
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568.
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447.
- Mor, D.E.; Murphy, C.T. Mitochondrial hyperactivity as a potential therapeutic target in Parkinson’s disease. Transl. Med. Aging 2020, 4, 117–120. Mor, D.E.; Murphy, C.T. Mitochondrial hyperactivity as a potential therapeutic target in Parkinson’s disease. Transl. Med. Aging 2020, 4, 117–120.
- Chen, M.; Vincent, J.; Ezeanii, A.; Wakade, S.; Yerigenahally, S.; Mor, D.E. Heparan sulfate proteoglycans mediate prion-like alpha-synuclein toxicity in Parkinson’s in vivo models. Life Sci. Alliance 2022, 5, e202201366.