1. Background, Composition, and Mechanism of Function
There has been evidence to support the hypothesis that some proteins can be ubiquitinated and degraded by the UPS by recruiting an E3 ligase in a way that is
proteolysis-targeting chimera (PROTAC
)-dependent ever since the discovery of PROTACs in 2001
[1][2][11,12]. Since its creation, PROTAC technology has found widespread application in both the development of novel therapeutics and fundamental biological studies. Over time, the application of the PROTAC technology has benefited a variety of distinct protein categories, including nuclear receptors
[3][13], kinases, G protein-coupled receptors (GPCRs)
[4][14], trans-membrane proteins, small GTPases, epigenetic proteins
[5][15], transcription factors, and protein aggregates
[6][16]. A linker connecting an E3 ubiquitin ligase (E3)-recruiting ligand and a protein gives a PROTAC molecule its dual function
[7][8][17,18].
2. Proteolysis-ROTargeting ChimeraACs: Targeted Management Strategy for Several Diseased Conditions
2.1. Proteolysis-Targeting Chimeras in the Cancer Treatment
2.1. PROTACs in the Cancer Treatment
The development of better cancer treatments is a priority for the medical community. Recent years have seen the development of innovative cancer therapeutic approaches, including CRISPR/Cas9 technology, RNA interference methods, antisense oligonucleotides, monoclonal antibodies, and small-molecule inhibitors (SMIs). CRISPR/Cas9 technology can only be used as a candidate technique, though, because the off-target effect is still a problem. Only a limited portion of ASOs now have FDA marketing approval
[9][58] due to a number of difficulties with RNA interference techniques and ASO medicines, including biological instability, immunogenicity, cell transport, and biological distribution
[10][77]. Antigen-specific binding, which reduces cell toxicity, is the idea behind monoclonal antibodies. However, their inability to target intracellular proteins or receptors along with their high cost precludes their application. Antigen-specific binding is the idea of monoclonal antibodies, which helps reduce cell toxicity. They cannot, however, target intracellular proteins or receptors, and their high cost precludes their use in medicine
[11][48].
Despite the present widespread use of small-molecule modulators that target tumor cells, there are still several downsides, including drug resistance, toxicity, and poor selectivity. Finding new treatment approaches is essential to overcoming the downsides of current drugs. The development of PROTAC has generated a lot of anxiety
[12][78]. This approach effectively avoids the problem of medication resistance by selectively destroying pathogenic proteins via the UPS, a unique intracellular protein degradation mechanism. Recently, various research groups have focused on PROTAC technology and have produced a large number of small PROTAC compounds for a variety of malignancies, including many cancers
[13][44]. The “De-Linker” unique graph-based deep generative model, which combines 3D structural data with cutting-edge machine learning techniques for scaffold hopping and fragment linking, was recently published by certain teams. As a result, as seen in various examples, it can also be used while designing PROTACs. Additionally,
rwe
searchers believe that in the future, the application of computational generative approaches in drug discovery will increase
[14][79]. A few PROTAC compounds’ IC
50 values can be as low as 1 nM in certain contexts.
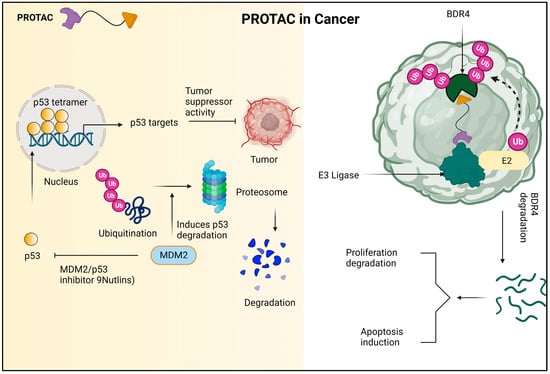
In addition to its superior ability to break down proteins, PROTAC has the ability to accelerate tumor regression, decrease tumor development, and create advanced anticancer effects on the cellular level. The mechanism of PROTAC against cancer cells is shown in Figure 1.
Figure 1. Due to the encouraging the ubiquitination and subsequent proteasomal destruction of the tumor suppressor protein p53, MDM2 has been identified as a possible target for anti-cancer therapy. This procedure is interfered with by nutlins and other MDM2/p53 interaction inhibitors, which prevent p53 from being degraded and instead cause it to accumulate. In turn, this strengthens p53′s anti-tumor properties and offers a viable plan for the creation of novel cancer treatments. The PROTAC molecule known as dBET1 is made up of two parts: thalidomide, which attracts the E3 ubiquitin ligase CRL4 CRBN, and JQ1, which binds to the oncoprotein BRD4. Using the cellular apparatus of the cell, dBET1 is designed to target BRD4 for breakdown by the proteasome. 7.2. PROTACs in neurodegenerative diseases.
In neurodegenerative diseases, which can worsen over time and lead to malfunction, neurons or their myelin sheaths are destroyed. Cerebellar atrophy, Parkinson’s disease, Huntington’s disease, and Alzheimer’s disease (AD) make up the majority of them clinically. Alzheimer’s disease (AD) is one of the most common neurological disorder and its clinical symptoms mostly include cognitive decline, behavioral abnormalities, and a reduction in functional capacity
[15][80]. The causes of AD are complex and comprise several variables. The pathogenic tau protein and the -amyloid (A) cascade are among those that have been intensively studied; however, recent findings suggest that the latter is more likely to be the focus of treatment for AD
[16][75], given that tau proteins are degraded by the peptide-based PROTAC TH006
[17][60]. An E3 ligand and a tau were the primary additions to the fusion peptide molecule. PROTACs, which stand for PROteolysis TArgeting Chimeras, have emerged as a promising therapeutic approach since their discovery in 2001. One of the main advantages of PROTACs is their ability to target and degrade proteins that were previously considered “undruggable,” making them a potentially important tool in treating a variety of diseases. In particular, in vitro studies have shown promise for using PROTACs as a therapeutic modality in triple-negative breast cancer (TNBC). Although the data suggests great potential, further research is needed, including in vitro and in vivo clinical trials, to fully establish the safety and efficacy of using PROTACs in TNBC treatment
[18][81]. PROTACs have emerged as a promising class of therapeutics for lung cancer and drug resistance in recent years. Researchers have developed several PROTAC molecules targeting validated therapeutic targets in NSCLC such as EGFR, KRAS, ALK, BRAF, and BCL-XL. These compounds have shown antitumor efficacy in cell models and preclinical tumor models. The development of PROTACs is rapidly advancing, and they hold great potential as novel therapies for lung cancer
[19][82]. PROTACs have been the subject of research for the past two decades, but only a few have demonstrated selectivity towards tumor cells. This is because many PROTACs recruit E3 ligases that are expressed ubiquitously in both normal and tumor tissues, which can lead to on-target toxicities. However, researchers are exploring various strategies to enhance the selectivity of PROTACs for tumor-specific proteins of interest (POIs) to improve their therapeutic potential
[20][83].
2.2. Proteolysis-Targeting Chimeras in Immune System Diseases
2.2. PROTACs in Immune System Diseases
Autoimmune disorders develop when the body’s immune response to self-antigens damages its own tissues. They can be added to by using acquired immunological disorders and are characterized by different levels of molecular and tissue damage, even organ failure and death
[21][84]. Genes, the environment, and risky habits such as smoking all have a role in the etiology of autoimmune illnesses. Despite extensive study and significant progress,
rwe
searchers still need to completely understand the mechanism and make additional efforts
[22][85]. IRAK4 activation has been associated with autoimmune diseases such SLE, psoriasis, rheumatoid arthritis, and cancer
[23][86]. As a result, Harling and his colleagues recently published a PROTAC molecule to target and destroy IRAK4
[24][87]. The authors of this
wor
esearchk successfully predicted the ideal role to bind IRAK4 ligands using molecular docking analysis based on the crystal structure of IRAK4 along with its kinase inhibitors. They then conducted a search on the constructed model and identified 12 atoms to be an appropriate duration of linker. General control non-derepressible 5 (GCN5) and P300/CBP-associated factor (PCAF), two related epigenetic proteins, can lower immune factors such as IL-6 and TNF that are caused by genes, the environment, and unhealthy habits such as smoking
[25][88].
ResWe
archers still need to completely understand the mechanism and make more effort despite extensive research and significant advancements
[22][85].
2.3. Proteolysis-Targeting Chimeras in Viral Infection
2.3. PROTACs in Viral Infection
The hepatitis B virus (HBV) mostly causes acute and chronic hepatitis B. The health of the entire world’s population is already at risk from HBV because millions of people are affected
[26][89]. These HBV-infected patients will eventually develop mild or light hepatitis, liver cirrhosis, and liver fibrosis, which will progress to hepatocellular cancer. Therefore, it is believed that HBV is the main cause of HCC
[27][90]. The two primary drugs used to treat HBV infection nowadays are reverse transcriptase inhibitors and interferons. However, neither of them makes a major difference in the fight against HBV. To fully address the issue of HBV infection, there is still considerable effort to be carried out. The X-protein trans-activator of HBV controls apoptosis, transcriptional networks, DNA repair networks, and intracellular signal transduction pathways. The X-protein trans-activator of HBV controls apoptosis, transcriptional networks, DNA repair networks, and intracellular signal transduction pathways
[28][91].
Studies have shown that the pathogenesis of HCC caused by HBV depends on the X-protein. It is clear that the X-protein promotes intracellular transcription and replication of HBV while blocking hepatic molecular apoptosis, hence promoting the growth of liver cancer cells. Apoptosis and HBV transactivation may be prevented by the X-protein’s ubiquitin-binding, unstable, and oligomerization regions, according to a previous publication. In 2014, a number of peptide-based PROTAC compounds with an emphasis on the X-protein were developed
[29][92].
The N-terminal oligomerization area and the C-terminal instability region of PROTAC molecules were joined by fusion of the X-N-proteins and C-terminal unstable sections, resulting in the formation of two distinct types of PROTAC molecules. Additionally, they were able to create several fusion peptides by replacing the C-terminal dangerous region with the oxygen-structured degradation (ODD) region of hypoxia-inducible factor (HIF-1), which interacts with VHL-type E3 ligase
[30][93]. Due to the previous ligand, they had been equally effective at destroying PROTAC, proving that the C-terminal’s dangerous area can also be utilized as an E3 ligase ligand. The potential for the PROTAC compounds to prevent or treat HBV infection and/or HCC should be investigated in preclinical studies.
2.4. Proteolysis-Targeting Chimeras in Metabolic Disease
2.4. PROTACs in Metabolic Disease
Molecular docking technology, fortunately, provides a speedy and efficient means to complete jobs and can greatly reduce prior preparation labor. A useful resource is the ability of molecular docking techniques to automatically discover putative binding sites between targets and ligands for some unidentified proteins
[31][94]. PNPLA3 (patatin-like phospholipase domain-containing protein 3) and prenyl-protein chaperone PDE are two examples of studies devoted to learning more about particular unknown proteins
[32][95], and PROTAC molecules have the capacity to degrade those target proteins. PROTAC technology, on the other hand, is a state-of-the-art method that can assist in obtaining profound insights into the mechanisms and functions of unknown proteins. Numerous studies have shown that PROTAC technology has the ability to continually influence with advanced discoveries that enhance the science of drug discovery.
3. Therapeutic Application of Proteolysis-Targeting ROTAChimera
Proteolysis-targeting chimeras (PROTACs) have fundamentally altered the way that medications are created due to their numerous advantages over traditional small-molecule inhibitors. As opposed to occupancy-driven inhibitors, PROTACs’ mechanism of action (MOA) is event-driven and catalytic in nature, leading to a stronger and longer-lasting effect. Furthermore, PROTACs offer a further level of selectivity that reduces potential toxicity and boosts effectiveness in the face of drug-resistance mechanisms. By concentrating on non-enzymatic processes, they can increase the range of potential treatment targets.
Since its discovery twenty years ago, PROTACs have developed from cell-impermeable peptide–small-molecule hybrids to clinical candidates that are orally bioavailable and can break down oncogenic proteins in people. The pace of scientific advancement is expected to quicken as
researchwe
rs approach the third decade of targeted protein degradation (TPD). The creation of ligands for previously “undruggable” proteins and the recruitment of new E3 ligases are made possible by advances in technology. Furthermore, improved computational power is assisting in the logical design of more powerful and selective PROTACs as well as the discovery of active degraders
[33][113]. When opposed to conventional inhibitors, PROTACs, also known as proteolysis-targeting chimeras, offer a novel pharmacodynamic strategy with a number of potential advantages. The ability to achieve pharmacodynamic efficacy even when the PROTACs are not detectable in the body is one of these benefits
[34][114]. Selectivity, effective distribution, and sensitivity to drug resistance are just a few of the advantageous characteristics of PROTACs. These features can be enhanced by using targeting ligand methods. These compounds function by selectively stimulating intracellular proteolysis, which has been shown to be effective in inhibiting cancer cell proliferation and encouraging apoptosis
[35][115]. A particular class of molecule known as PROTAC can benefit from the advantageous interaction between two amyloid proteins. The PROTAC-induced close proximity of the amyloids allows for the formation of a stable ternary complex, which can boost the cross-interaction’s beneficial effects. The PROTAC molecules will be built using peptide mimics with a high affinity for a 1-42
[36][37][116,117].
Alzheimer’s disease and related tauopathies are characterized by tau buildup within cells, and targeting tau has emerged as a promising strategy for therapeutic development. In order to selectively degrade proteins inside of cells, the proteolysis-targeting chimera (PROTAC) approach was developed. In order to achieve this, a novel small-molecule PROTAC with the designation C004019 and a molecular mass of 1035.29 dalton was developed. PROTAC was created to specifically increase tau protein ubiquitination and proteolysis by recruiting tau and E3-ligase (VHL)
[38][118]. The first VHL-based small-molecule PROTAC designed to target nuclear hormone receptors was called PROTAC-ERR. These PROTACs have a lot of medicinal promise
[39][119]. A substance called PROTAC-ERR can aim to degrade estrogen-related receptor alpha. The orphan receptor estrogen-related receptor alpha (ERR), which is found in the nucleus of MCF-7 breast cancer cells, can be targeted for degradation by the substance PROTAC-ERR.
At a dose of 100 nM, PROTAC-ERR can cause these cells to degrade approximately 50% of ERR. The tyrosine kinase family of enzymes, which JAKs are a member of, is involved in the transmission of cytokine-mediated signals in cells. JAKs phosphorylate tyrosine residues when they are activated, which can subsequently activate downstream signaling proteins and cause a variety of physiological effects. These enzymes are able to transmit information from external chemicals such as cytokines, growth factors, and chemokines to the cell’s nucleus, where they can directly affect DNA transcription and the translation of a number of proteins. The highlighted paragraph discusses how various JAK proteins express themselves differently in various cell types. The widespread expression of JAK1, JAK2, and TYK2 contrasts with the predominant presence of JAK3 in hematopoietic, myeloid, and lymphoid cells. The patent also lists a number of JAK2-binding PROTAC substances, such as ruxolitinib and baricitinib, which work by targeting JAK2 JH1 in people. In MHHCALL-4 cells, the substances were examined for their capacity to cause protein degradation, cytotoxicity, and effects on the JAK-STAT signaling pathway
[40][120].
Protein kinases—which are often mutated in the human genome and involved in cellular processes such as cell apoptosis, signaling, signal transduction, and immune-response propagation—are linked to cancers. Both IL-1R and TLR dimerize as a result of ligand binding, and adaptor molecules are recruited to a highly conserved toll/IL-1R (TIR) domain on the cytoplasm. E3 ubiquitin ligases have received increased attention recently in drug development attempts because they are more desirable therapeutic targets. MDM2 inhibitors, which target the E3 ligase mouse double minute 2 homologue (MDM2), and von Hippel–Lindau (VHL) tumor suppressor, which is the substrate recognition component of the E3 ligase complex VCB, are two examples of specific ligands that have been developed to bind to these ligases. By responding to DNA damage or stress and controlling cell development, the tumor suppressor gene p53 also exerts a significant influence on apoptosis
[41][121].
A possible method for causing tailored protein degradation is the proteolysis-targeting chimera (PROTAC) technology. In this strategy, heterobifunctional molecules are used to attract an E3 ubiquitin ligase to a particular protein of interest, causing the proteasome to degrade it. Indirectly affecting upstream signaling cascades, transcriptional programs, or epigenetic processes can result in PROTAC-mediated protein degradation. This technology has proven to be successful in numerous preclinical and clinical investigations, demonstrating its potential as a cutting-edge treatment strategy
[42][122]. A promising small-molecule therapy approach for treating disorders connected to the androgen receptor (AR), such as prostate cancer, Kennedy’s disease, and cardiovascular conditions, is the PROTAC idea. The capacity of PROTACs to lower protein levels is one of its main benefits. When compared to conventional small-molecule inhibitors, this property allows for small-molecule degraders to achieve a more comprehensive target inhibition, which could lead to more effective treatments
[43][123].
A prospective target for the therapy of numerous disorders, including cancer, is cyclin-dependent kinases (CDKs). Compound iCDK9, a highly selective CDK9 inhibitor, is one possibility. The possible toxicity of this inhibitor and
our incomplete understanding of its mechanism, however, pose some limits. A class of bioactive molecules known as PROTAC (proteolysis-targeting chimeras) degraders can selectively stimulate the degradation of their target protein in vitro and in vivo, therefore lowering the dose-limiting toxicity of small-molecule medications
[44][124]. Ao et al. (2023) recently developed and synthesized
[44][124] bifunctional PROTAC compounds to target iCDK9 and show its hitherto unidentified target and pharmacological mechanism. The CD-5 chemical showed minimal toxicity in cells while selectively degrading CDK9. While CRBN-based PROTACs such as ARV-110 and ARV-471 have drawn a lot of attention for their therapeutic potential in treating cancer and other diseases, the study emphasizes that they have also been thoroughly investigated throughout the world and have been found to be effective in treating a variety of illnesses including viral infections, cardiovascular diseases, immune disorders, and neurodegenerative diseases
[45][125].
Histone deacetylase 6 (HDAC6) may be a therapeutic target for the treatment of a number of disorders, according to a recent study. According to the study, since HDAC6 is essential for the activation of the NLRP3 inflammasome, targeting it may be useful in the treatment of inflammatory illnesses. In order to accomplish this, Cao Z et al. (2021) created an HDAC6 degrader with minimal cytotoxicity using the PROTAC approach, which targets proteolysis. They did this by combining pomalidomide, a CRBN E3 ligand, with a selective HDAC6 inhibitor produced from the natural product indirubin. Multiple cell lines, including active THP-1 cells, had their HDAC6 levels efficiently and arbitrarily decreased by the HDAC6 degrader
[46][126]. Similarly to antibody–drug conjugates (ADCs), antibody–PROTAC conjugates have become a possible method for the selective administration of a broad-spectrum PROTAC to particular cell types. Due to their capacity to deliver cytotoxic drugs to cancer cells only, ADCs have become more and more prominent in the treatment of cancer
[47][127].
He and colleagues created the aptamer–PROTAC conjugate (APC) in their 2021 study by affixing a BET-targeting PROTAC to the nucleic acid aptamer AS1411 (AS) with a cleavable linker. By improving the molecule’s (APR) capacity to target tumors in an MCF-7 xenograft model, this design also reduced toxicity while enhancing BET degradation and anti-tumor effects. The researchers used this method to improve the molecule’s distribution and selectivity, highlighting its potential for use in medicinal applications in the future
[48][128].
Proteolysis-targeting chimeras (PROTACs) have become a promising approach for investigating pharmacological targets that are difficult to target with conventional methods. In recent years, these chimeric molecules have shown effective targeting capabilities, overcoming the challenges posed by traditional approaches. With conventional therapies, medication resistance is frequently unavoidable; nevertheless, this unique technique offers a means of overcoming it. In order to combat acquired drug resistance, PROTACs offer a potentially effective approach by promoting the targeted degradation of particular proteins.