Dilated cardiomyopathy (DCM) represents one of the most common causes of non-ischemic heart failure, characterised by ventricular dilation alongside systolic dysfunction. Despite advances in therapy, DCM mortality rates remain high, and it is one of the leading causes of heart transplantation. Developments in complementary diagnostic procedures, namely cardiac magnetic resonance and genetic testing, have shed new light on DCM understanding and management.
- dilated cardiomyopathy
- risk stratification
- cardiovascular magnetic resonance
1. Introduction
2. Diagnostic Workup
Considering the broad spectrum of disorders that cause DCM, a systematic approach helps to identify and manage this condition, especially the more uncommon but clinically significant forms of DCM. A dedicated diagnostic workup (Figure 1) will lead to an aetiology-oriented approach to help with risk stratification and advice on therapeutic interventions.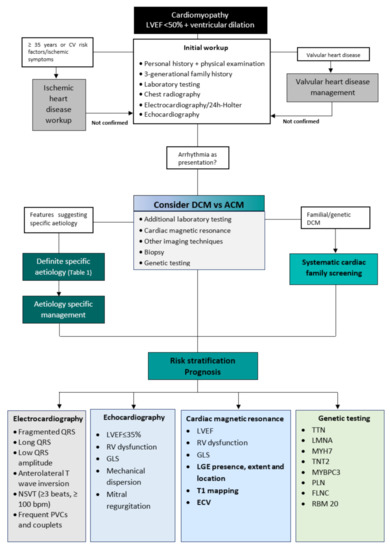
3. Differential Diagnosis
In the differential diagnosis of DCM, it is mandatory to exclude the most frequent causes of LV dysfunction, such as coronary artery disease and valvular disease, where LV dilatation can occur due to adverse remodelling. The exclusion of epicardial coronary artery stenosis is carried out using invasive coronary angiography, the gold standard, or by computerised tomography in patients not at high risk of atherosclerosis. Valvular disease should be systematically sought through various imaging methods [6]. Other less-common cardiomyopathies can have phenotypic similarities with DCM. They must also be considered in the differential diagnosis, such as left ventricular non-compaction cardiomyopathy (LVNC) and peripartum cardiomyopathy (PPCM). LVNC is an unclassified cardiomyopathy that, in advanced cases, can present findings similar to DCM (dilated and impaired LV function) [10]. Echocardiographic findings compatible with LVNC consist of prominent trabeculations, and deep recesses found mainly in the LV’s apex and free wall and a two-layer wall structure, with an end-systolic ratio of >2 between the non-compact subendocardial layer and the compact subepicardial layer [10][11][10,11]. In addition, imaging with cardiac magnetic resonance (CMR) may show the extent of myocardial involvement and the degree of fibrosis with late gadolinium enhancement (LGE) [11]. PPCM is an idiopathic cardiomyopathy with HF, secondary to LV systolic dysfunction, usually seen in the last months of pregnancy or the first five months after delivery. A reduction in LVEF is required to establish the diagnosis, but the left ventricle may or may not be dilated [12][13][12,13]. Despite the distinct classification, DCM and LV hyper trabeculation often overlap due to LV remodelling, and DCM and PPCM may share the same genetic background [13]. Findings of extracardiac manifestations, namely neuromuscular involvement, should alert for other rarer syndromic or metabolic cardiomyopathies [14].Differential Diagnosis of Dilated and Arrhythmogenic Cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM) is a genetic heart muscle disease characterised by the replacement of the ventricular myocardium with fibrofatty tissue. Although initially defined as a condition that distinctively affected the right ventricle (RV), more recent post mortem and contrast-enhanced CMR investigations revealed that the left ventricle is often involved [15][16][15,16]. ACM is distinguished from DCM by a propensity towards arrhythmia exceeding the degree of ventricular dysfunction. However, there is a subgroup of DCM patients with clinical presentations of arrhythmia and syncope early in the disease course [15][16][17][15,16,17]. The term ‘arrhythmogenic DCM’ has been used to describe these patients. This subset of arrhythmogenic DCM patients shows genotypic and phenotypic features that overlap with those of arrhythmogenic left ventricle cardiomyopathy (ALVC): more frequent arrhythmic events, LV systolic dysfunction, mild LV dilatation, and myocardial fibrosis. Consequently, the differential diagnosis between DCM and ALVC may be challenging, but it is essential, due to their different management and prognosis [16][17][18][16,17,18]. Conduction system disease and frequent atrial or ventricular arrhythmias (VA), especially with a near-normal LVEF, should raise suspicion of ACM [16]. Low voltages at limb leads, inverted T waves in V4-V6, frequent premature ventricular contractions (PVC), and non-sustained ventricular tachycardia (NSVT) of right bundle branch block morphology are findings suggestive of ALVC [16][17][19][16,17,19]. Therefore, a 24-h Holter monitoring should be performed on every patient suspected of ACM. The presence of >1000 PVC or NSVT should raise concerns about the propensity for arrhythmogenicity [16]. While the typical phenotype of DCM consists of LV dilatation with systolic dysfunction, the ALVC remodelling pattern may present with a normal or only slightly depressed LV function [16][20][16,20]. In distinguishing both entities, an imaging study using CMR is fundamental. While LGE is detected in less than half of DCM cases, nearly all patients with ALVC with LV systolic dysfunction show the presence of LV LGE [21][22][21,22]. The distribution of LGE differs between the two conditions: in ALVC, it is predominantly distributed in the subepicardial inferolateral regions; in DCM, it usually affects mid-mural septal segments [21]. Fatty myocardial infiltration may be assessed in dedicated sequences in the CMR study, and is often observed in the same regions of LGE. LV myocardial fibrosis/LGE is significantly higher in patients with ALVC than those with DCM [22][23][22,23]. It is known that while the LGE in DCM represents an epiphenomenon and is unrelated to the reduction of the LV systolic function, in ALVC, it is directly correlated with the degree of LV dysfunction [22][23][24][22,23,24]. In more advanced stages of ALVC, fibrofatty tissue can deposit in multiple segments of the LV free wall and septum, with transmural involvement, leading to severe systolic dysfunction [20]. Demonstrating an ACM-causing gene mutation associated with a consistent phenotype is mandatory for diagnosing ALVC [16]. The predominant genetic background in ALVC includes the mutations of genes encoding for desmosomal proteins, lamin A/C, phospholamban, filamin C, RMB20, and SCN5A. In DCM, the most frequent genes involved encode cytoskeleton, muscular sarcomere, and nuclear envelope proteins. Nonetheless, significant genetic overlap exists [17][23][25][17,23,25].4. Aetiologies
DCM is a heterogeneous disease encompassing various underlying causes, including genetic and acquired disorders. A positive family history can be detected in up to 30–50% of DCM cases, and a causative genetic mutation can be identified in up to 40% of DCM cases [2][6][26][2,6,26]. In the presence of positive cases in the family, sarcomeric, neuromuscular, and mitochondrial disorders are the most frequent aetiologies. External factors such as exposure to toxins, diabetes, arrhythmia, myocarditis, and pregnancy often contribute to the development of the phenotype and outcome [26]. The non-genetic causes of DCM include infectious (viral or non-viral), autoimmune, toxic, infiltrative-related causes, nutritional deficiencies, and endocrine disorders [26].
5. Risk Stratification and Prognosis
Despite advances in DCM treatments, 10-year survival remains less than 60%, with death preceded by numerous HF exacerbations. Remarkably, the clinical course of DCM patients varies widely, ranging from rapidly progressive HF or SCD to LVRR, denoting high complexity in assessing the individual risk [2][4][5][2,4,5]. Various elements from clinical presentation, previous medical history and comorbidities, physical examination, and laboratory and imaging investigations are available, to enhance risk stratification.5.1. Electrocardiogram
A fragmented QRS, longer QRS duration, antero-lateral T-wave inversion and low QRS voltage in the ECG are associated with a higher risk of major arrhythmic outcomes, including death due to arrhythmia, appropriate implantable cardioverter-defibrillator (ICD) therapy, documented VT/VF, and all-cause mortality [27]. Previous studies showed that longer QRS (>120 ms), together with LGE on CMR, provided incremental value to LGE alone in predicting all-cause mortality [28]. The 24-h Holter monitoring demonstrates NSVT in 40–60% of patients and polymorphic PVC in up to 90% of DCM patients. The presence of NSVT and frequent PVC (≥1000 PVC or ≥50 couplets/24 h) increases the arrhythmic risk, mainly when combined with a family history of malignant VA or SCD [29][30][29,30].5.2. Echocardiography
Imaging with echocardiography is not only indispensable in diagnosing DCM, but it also provides multiple prognostic indicators. LV systolic function is one of the most critical evaluations. It has been considered the primary determinant of prognosis, guiding patient management and subsequent treatment, including the indication for ICD, cardiac resynchronisation therapy (CRT), or the discontinuation of cardiotoxic chemotherapy. For a more accurate LV function assessment, it is recommended that a 3-dimensional (3D) echocardiography should be performed to determine LVEF, when available in experienced laboratories, as it can overcome some of the limitations inherent to a 2-dimensional (2D) LVEF measurement [31][32].5.3. Cardiac Magnetic Resonance
5.3.1. Late Gadolinium Enhancement
Myocardial fibrosis occurs due to collagen accumulation resulting in interstitial expansion without myocardial necrosis (interstitial fibrosis) and from cardiomyocyte death (replacement fibrosis). There is a good histological correlation between LGE and replacement fibrosis, but LGE has a low sensitivity for interstitial fibrosis [32][40]. Late gadolinium enhancement is present in around 30% of patients with DCM, typically in a mid-wall pattern. It has been associated with the occurrence of major VA consistently across all subgroups, regardless of LVEF. Besides the association with a four-fold increased risk of SCD or aborted SCD, LGE is an independent predictor of cardiovascular mortality and HF hospitalisation. Prognostic value is added with LGE beyond LVEF, NHYA class, and common baseline cardiovascular covariates [32][33][34][40,41,42].5.3.2. T1 and Extracellular Volume
The study of interstitial fibrosis using T1 mapping and extracellular volume (ECV) estimation is an expanding area of CMR. Techniques for calculating T1 mapping have shown a good correlation with histologically graded myocardial fibrosis and, as such, may allow for a more sensitive detection of the early disease process, and initiation of respective treatment before overt pathology [35][46]. In addition, a higher native-T1 value of myocardium constitutes an independent predictor of all-cause mortality and HF events in patients with DCM [36][47], and is also a predictor of arrhythmic outcomes, including the appropriate ICD therapy or sustained VA [37][48]. Extracellular volume has been shown to hold prognostic value incremental to LGE or native-T1 mapping [38][49]. A strong association has been demonstrated between ECV and major adverse cardiac events, including heart failure hospitalisations and all-cause mortality [37][38][48,49]. Abnormal ECV measurements yield a 2.8-fold increased odds of negative outcomes, independently of age, sex, functional class, and LVEF [38][49]. Patients with DCM and a high ECV and prolonged QRS duration had a significantly worse prognosis than those with normal ECV and QRS duration [37][38][48,49].5.3.3. Feature-Tracking Strain Analysis
Feature-tracking (FT) strain analysis is a promising tool for improving DCM patients’ risk stratification. Evidence suggests that survival prediction and risk stratification in DCM may be strengthened by FT parameters, independently of clinical parameters, biomarkers, LVEF, and LGE. For example, a preserved GLS determined by CMR carried a good prognosis, even in patients with LVEF < 35% and those with LGE [39][50].5.4. Genetic Testing
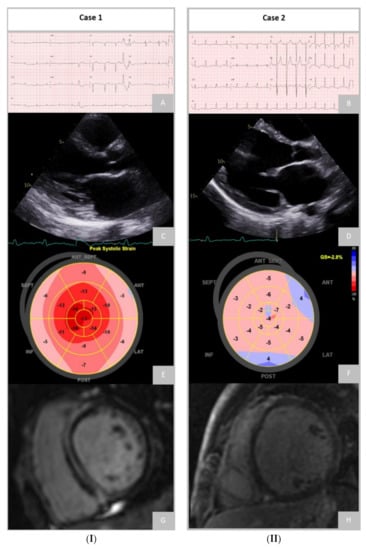
DCM has a recognisable genetic background that significantly overlaps with other cardiomyopathies, which could partially explain its wide heterogeneity. Current evidence suggests that 30% to 40% of DCM cases are caused by pathogenic or likely-pathogenic gene variants that occur in more than 40 genes associated with the condition [14][40][41][14,51,52]. Genetic testing can influence clinical management in patients with DCM, as once a mutation is identified and its pathogenic role and mode of inheritance are established, that information can be used for guidance of therapy and family screening. Figure 2 demonstrates the remarkably different phenotype and diagnostic findings that can be found in patients with different genetic mutations.