The development of antiretroviral drugs (ARVs) was a great milestone in the management of human immunodeficiency virus (HIV) infection. ARVs suppress viral activity in the host cell, thus minimizing injury to the cells and prolonging life. However, an effective treatment has remained elusive for four decades due to the successful immune evasion mechanisms of the virus. This review discusses the HIV-host interactions to help us understand the mechanisms involved.
- HIV
- AIDS
- immunity
- cells
- T- cell exhaustion
- CCR5
- CXCR4
- sex
1. Introduction
Tremendous progress in the understanding of the HIV molecular interaction with the host cell, the host cell responses to the virus and potential therapeutic implications of this interaction has been made since its discovery [30][31]. Vigorous research heralded the development of antiretroviral drugs a very important milestone in controlling the HIV pandemic [32]. Despite much progress in understanding the HIV–host cell interactions, the cure for HIV infection has remained elusive for four decades now [31][33].
2. Structure of HIV
3. HIV Life Cycle
The HIV life cycle consists of 11 phases and includes binding/attachment, fusion, trafficking, nuclear import, reverse transcription, integration, transcription/translation, assembly, budding and release [38][39] (Figure 1).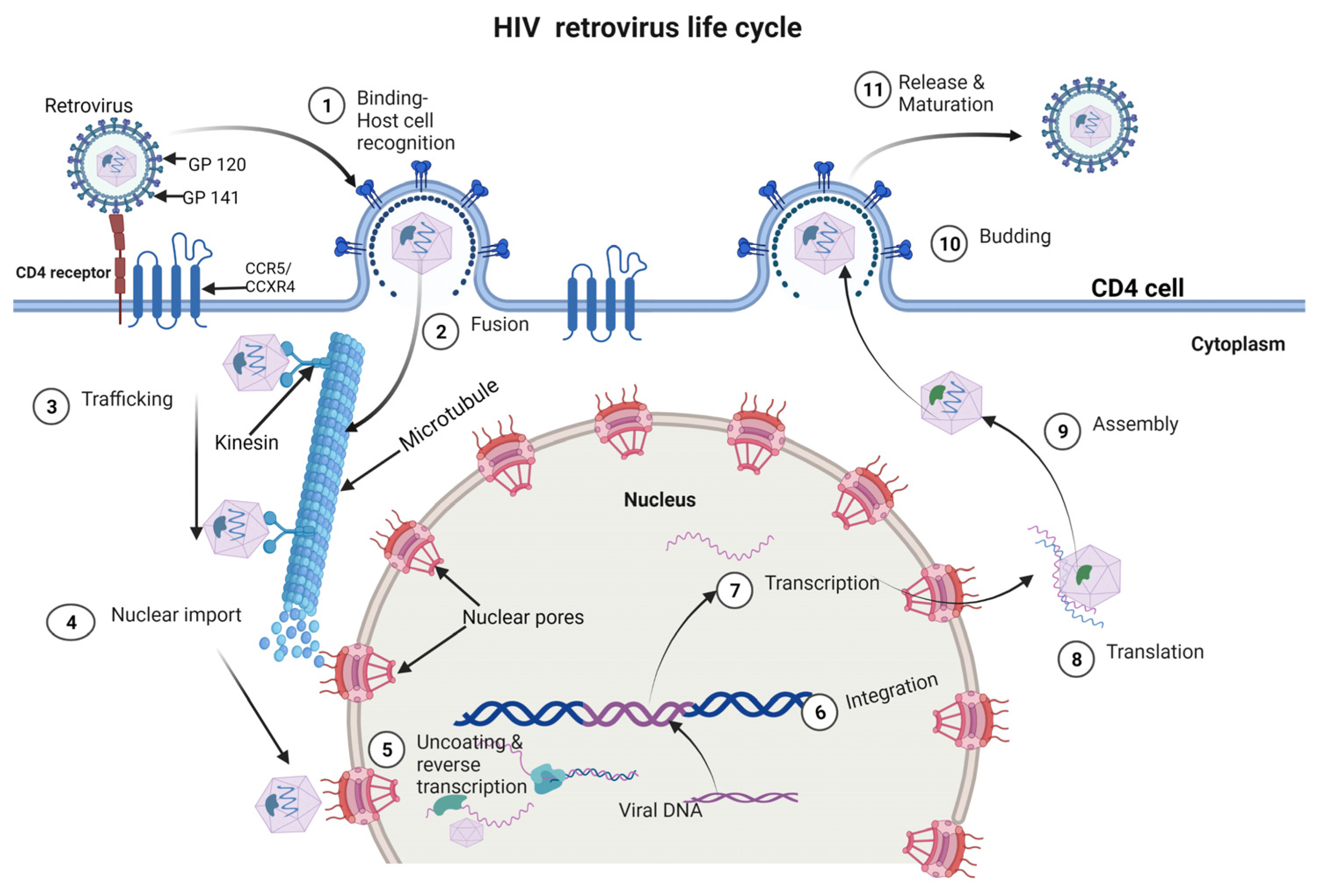
4. HIV-Related Factors Promoting Infection and Immune Evasion
4.1. Downregulation of MHC Class I and II
4.2. Production of Non-Neutralizing Antibodies
The viral envelope glycoprotein, gp120, is highly variable, and it can quickly mutate to escape recognition by neutralizing antibodies that target specific regions of the protein, thus leading to the production of non-neutralizing antibodies that can bind to gp120 but are unable to block virus entry [11][41]. Non-neutralizing antibodies can still play a role in HIV immune evasion. By binding to gp120, they can prevent the recognition of viral epitopes by neutralizing antibodies or T cells, effectively shielding the virus from immune surveillance [63]. Non-neutralizing antibodies can also trigger Fc receptor-mediated signaling, which can downregulate immune effector cells, such as Natural Killer (NK) cells and macrophages, leading to decreased antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis of infected cells [64][65].4.3. Induction of Immune Exhaustion
HIV can induce immune exhaustion, which is a state of functional impairment of T cells, at a molecular level by several mechanisms. First, persistent antigen stimulation caused by HIV infection leads to T cell activation and proliferation, eventually leading to T cell exhaustion [12]. Second, HIV upregulates inhibitory receptors on T cells, such as programmed cell death receptor 1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4) and T-cell immunoglobulin domain- and mucin domain-containing protein 3 (TIM-3), which negatively regulate T cell activation and function [12]. Third, HIV downregulates the expression of key transcription factors and cytokines, such as the T-Box protein expressed in T cells (T-bet), interferon-gamma (IFN-γ) and IL-2, that are necessary for effector T cell function [12][66].4.4. Destruction of Virus-Specific T Helper Cells
HIV can evade the host immune system by destroying virus-specific T helper cells, which are important for coordinating the immune response against the virus [67]. This occurs at a molecular level through several mechanisms [68]. First, HIV can directly kill infected T helper cells by inducing apoptosis or programmed cell death [69]. Second, HIV-infected cells can also cause the bystander killing of uninfected T helper cells through the release of viral proteins, such as Tat, Nef and gp120, which activate apoptosis pathways in nearby cells through several mechanisms such as the upregulation of Fas, FasL and TNFα expression [70], the reduced expression of Bcl-2 and the activation of p53 [71]. Third, HIV proteins can induce cell death pathways by disrupting the normal functioning of cellular proteins and organelles, such as the mitochondria, which can lead to the death of infected and uninfected T helper cells [67][72][73].4.5. The Emergence of Antigenic Escape Variants
HIV can evade the host immune system through the emergence of antigenic escape variants, which are viral strains that have mutations in the viral proteins that are recognized by the immune system [74]. Several mechanisms favor this. First, HIV replicates at a high rate, which results in the generation of a large number of viral particles that can potentially acquire mutations [75]. Second, the HIV reverse transcriptase, the enzyme responsible for copying the viral genome, is highly error-prone, which increases the likelihood of mutations occurring during replication [76]. Third, the immune system exerts selective pressure on HIV by targeting specific viral proteins, which can result in the emergence of variants that are less recognizable by the immune system [67].4.6. Expression of an Envelope Complex That Minimizes Antibody Access
HIV can evade the host immune system by expressing an envelope complex that minimizes antibody access, which refers to the outer surface of the virus that is recognized by the immune system [77]. The envelope protein of HIV undergoes molecular-level changes through various mechanisms. The envelope protein of HIV is covered in sugar molecules, making it highly glycosylated, and this can prevent antibodies from binding to and neutralizing the virus by shielding vulnerable regions of the envelope protein from antibody recognition [78].4.7. Dysregulation of the JAK/STAT Pathway
Interferons are primarily produced and released by host cells such as immune cells (macrophages, dendritic cells, T cells) and non-immune cells (fibroblasts, epithelial cells) in response to viral infections, certain bacterial infections or other immune triggers [79]. Upon detecting viral particles, the host production of interferons creates an antiviral atmosphere which suppresses viral replication through the mechanism of inducing the expression of antiviral proteins and activating immune cells [80][81][82][83].4.8. Other Factors That Promote HIV Infection
The HIV-1 viral infectivity factor (Vif) is a 23-kDa protein found within the HIV-1 virion that plays a crucial role in the survival/invasion of host tissue by HIV [84]. It counteracts the APOBEC3 family of proteins, which are host cellular defense mechanisms that can mutate the genetic material of viruses, including HIV. Vif targets Apolipoprotein B mRNA Editing Catalytic Polypeptide-like (APOBEC3) proteins for degradation, allowing the virus to continue to replicate and spread [85]. Without Vif, HIV is much less able to infect and replicate in host cells [85]. Additionally, the Elongin–Cullin–SOCS (ECS) box site is involved in several HIV-related factors that promote infection and immune evasion [86][87]. The HIV-1 Vif can interact with the ECS box site on SOCS proteins, leading to the dysregulation of cytokine signaling pathways and promoting viral replication and immune evasion [86][87]. The ECS box site is also involved in the regulation of interferon signaling pathways, and the dysregulation of these pathways by HIV can contribute to immune evasion and pathogenesis of the virus [86][88][89].5. Host Cell Mechanisms That Control Infection and Replication
HIV-1 infection progression is determined by both the virus and the host cells, with pattern recognition receptors (PRRs) playing a vital role in initiating the host immune response [90]. Early HIV-1 infection, the first hours to days after infection, in which the virus replicates in the cells such as the dendritic cells and macrophages/monocytes and is not detectable in the blood, is referred to as the “eclipse phase” [91]. The characteristics of early/recent infection include a high viral load and immune cell depletion. This eventually leads to immunodeficiency, and without treatment, individuals die of AIDS [92]. However, at the onset of infection, innate immune cells such as dendritic cells, NK cells, NKT cells, ϒδ T cells and B1 cells macrophage/monocytes respond to infection and also induce the cells of the adaptive immune system, the CD4+ and CD8+ T lymphocytes [93]. The innate immune response, requiring no gene rearrangement, is non-specific and uses pattern recognition receptors to recognize the HIV infection and induce other innate related factors against HIV [94]. These innate immune components include skin mucosal epithelial cells, phagocytes and NK cells, as well as a series of soluble factors, such as cytokines, chemokine and small molecular substances, such as complement and mannose-binding lectin [95]. The secreted IFNs produce an antiviral effect by autocrine and paracrine ligation to interferon-alpha/beta receptors (IFNAR) on cell surfaces [96]. This activates the downstream JaK/STAT signaling pathway through receptor-associated Jak1/TyK2 (tyrosine kinase) [97]. The phosphorylated STAT1 and STAT2 then form a heterodimer that interacts with IFN-regulatory factor 9 (IFR9) to form an IFN-stimulated gene factor 3 (ISGF3) transcription complex. ISGF3 translocate to the nucleus, where it binds to IFN-stimulated response elements (ISREs) in gene promoters, leading to the expression of IFN-stimulated genes to establish the host antiviral status that impairs viral replication and promotes the maturation of dendritic cells, promoting the activation of adaptive immune response [98]. Sentinel dendritic cells and macrophages are powerful, professional antigen-presenting cells that not only play a significant role in the initial response to infection but also activate adaptive immunity [95]. While sentinel dendritic cells are the first cells in response to infection, macrophages are the main effector cells involved in the late innate immune response and support the recruitment of inflammatory cells by secreting cytokines such as IL-1 and TNF-α [99]. Other cytokines, such as IFN-α and IL-15, which are secreted by dendritic cells and monocytes are significant in the activation of NK cells [100].5.1. Pathogen Recognition Receptors (PRRs)
Pathogen Recognition Receptors (PRRs) are immune receptors that recognize conserved molecular patterns on pathogens, such as bacteria or viruses [101]. PRRs are differentially expressed by various immune cells including macrophages and dendritic cells [102]. PRRs are an essential component of the innate immune system and initiate downstream signaling that leads to the production of cytokines, chemokines and molecules capable of activating the adaptive immune response [103]. PRRs can be divided into several classes, such as Toll-like receptors (TLRs), Nod-like receptors (NLRs), RIG-I-like receptors (RLRs) and C-type lectin receptors (CLRs), among others [104]. TLRs (Figure 2) are the most studied and characterized PRRs and recognize a broad range of pathogen-associated molecular patterns (PAMPs), including lipopolysaccharides, lipoproteins and viral nucleic acids [105]. Once bound to their specific ligands, TLRs activate multiple downstream signaling pathways, including the NF-kB pathway and the interferon regulatory factor (IRF) pathway, leading to the expression of genes that drive the inflammatory and antiviral immune response [106][107].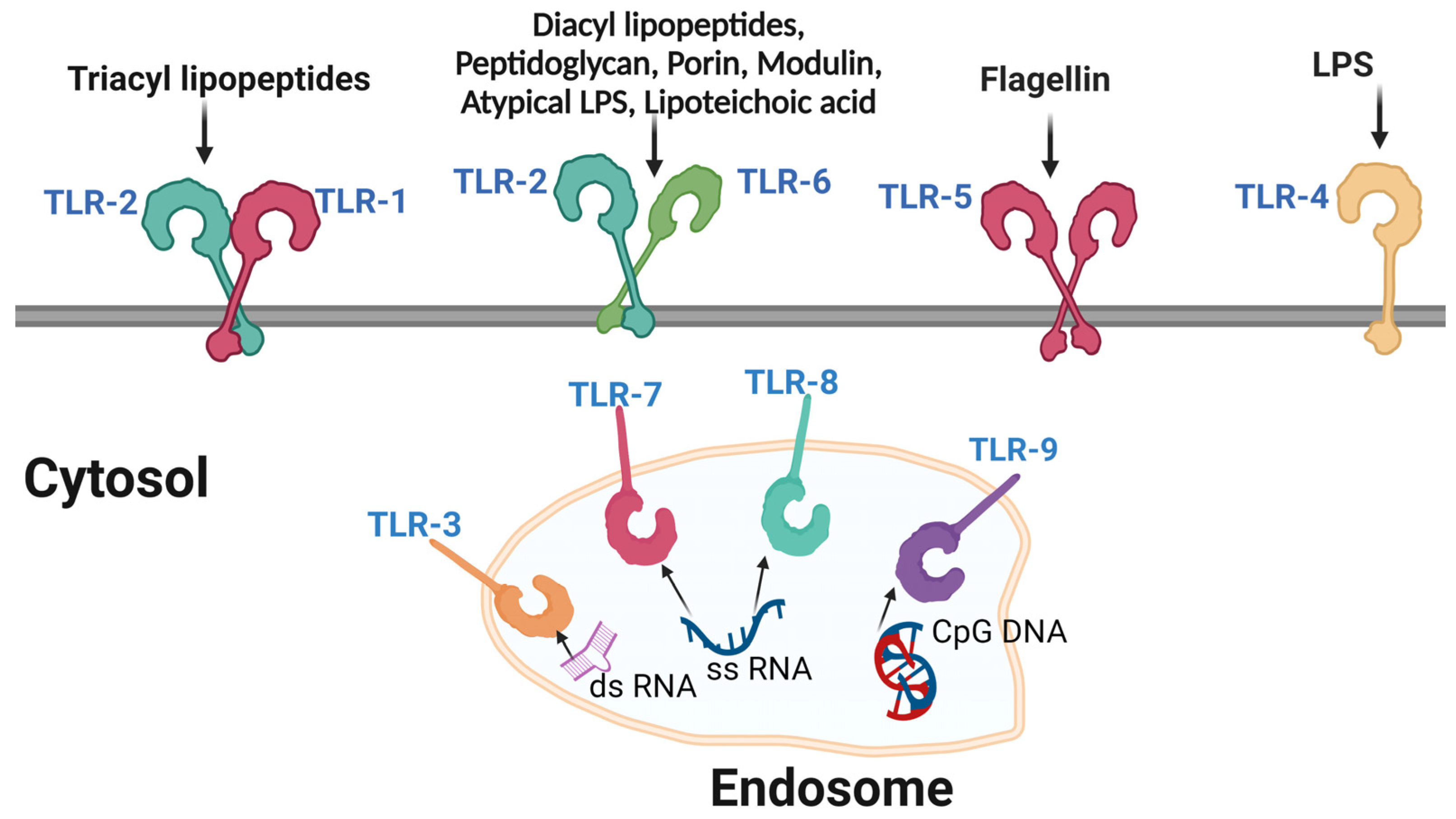
5.2. Dendritic Cells
5.2.1. Plasmacytoid Dendritic Cells (pDCs)
The most characteristic feature of the pDCs is the production of the type 1 interferon that promotes a strong antiviral immune response [110]. Plasmacytoid DC expresses TLR7, which enables it to recognize the virus after uptake by endocytosis and activate a signaling cascade that leads to the maturation of pDCs, the production of IFN-α, IFN-β and TNF-α and the expression of chemokine receptors such as the CCR5, CD40, CD80 and CD86 co-stimulatory molecules [94].5.2.2. Conventional Dendritic Cells (cDCs)
Conventional DCs function mainly as specialized APCs; however, they also produce several cytokines upon recognition of an antigen, mainly inflammatory cytokines including IL-6, IL-12, IL-15, IL-23, TNF and IL-1β all, of which are significant in restraining HIV-1 infection as compared to pDCs, which are known for the secretion of a large amount of type I interferons [93]. Conventional DCs are important in bridging innate immunity and adaptive immunity by presenting antigens to T cells [93]. Whereas cDCs1 are distinguished by their effective MHC class I-mediated priming of CD8+ T cells, cDC2 have a broad variety of factors generated and high cross-presenting abilities, promoting a potent activation of Th1, Th2 and Th17 as well as CD8+ T cell responses [93][95][99][109].5.3. Macrophages
Macrophages are key players in innate immune responses to pathogens, and their ability to destroy a wide range of pathogens while doubling as APCs makes them a vital component of the innate immune system [111]. Unlike most cells of the myeloid lineage, macrophages have a longer life span, ranging from months to years [112]. Macrophages are widely distributed in the body and reside in almost every tissue of the body [113]. While initially thought to be incapable of self-renewal, there is evidence that tissue macrophages can and do replenish themselves [114][115][116]. Viral interaction with macrophages is very important in the HIV disease course [117]. In the sexual transmission of HIV, macrophages encounter HIV in the genital mucosa along with CD4+ T cells and DCs [118][119]. Macrophages play a crucial role in the immune response to HIV infection in the early stages of the disease, as their primary function is to engulf and clear viral particles and infected cells [120][121].5.4. CD4
+
T Cells
CD4+ T cells are crucial components of the immune system and play a key role in mounting an effective response against viruses such as HIV [9]. However, HIV specifically targets and infects CD4+ T cells, leading to a gradual depletion of this cell population and ultimately resulting in the onset of AIDS [122]. The mechanisms underlying the CD4+ T cell response to HIV infection involve a variety of signaling pathways and biochemical interactions [123]. Upon the initial encounter with HIV, CD4+ T cells become activated and initiate a series of intracellular signaling events, including calcium flux and protein kinase C activation [84][124][125].5.5. CD8
+
T Cells
CD8+ T cells recognize and directly eliminate virus-infected cells. In the acute phase of HIV infection, there is an increase in CD8+ T cell activity due to APCs and CD4 T cell stimulation, resulting in CD8+ T cells killing virus-infected cells by releasing granzymes, which can induce apoptosis in the target cell and a pore-forming protein called perforin, which perforates the cell membrane of the target cell, thereby killing the cell [126]. CD8+ T cells recognize infected cells through the presentation of viral peptides on major histocompatibility MHC 1 molecules [127]. Once they encounter an antigen on MHC 1 molecules, they become activated, gain cytotoxic activity and additionally secrete a variety of cytokines including IFN-γ, which inhibit viral replication and create an antiviral environment [67][128].6. Influence of Sex on HIV Transmission and Immune Responses
There are notable sex differences in HIV infection transmission and progression. HIV infection in females is marked by a stronger initial immune response, characterized by a high CD4+ T cell count, low viral load and high CD8+ T cell activity, while infection in males is marked by high viral load, a lower CD4+ T cell count and low CD8+ T cell cytotoxic activity [129][130]. However, there is early immune exhaustion in females, accelerating the progression to AIDS at a rate comparable to that of males, and progression to AIDS occurs at a lower viral load compared to that of males [131]. Both the foreskin and the vaginal mucosa contain CD4+ T cells, which can be infected by HIV during sexual intercourse with an infected person; however, male CD4+ T cells express higher CCR5 receptors when compared to females, which implies that males are more likely to be infected in one sexual encounter with higher viral particles, thus partially explaining the higher viral loads in males in primary HIV infection [132][133]. Furthermore, the Langerhans cells in the vagina and foreskin transport viral particles to the local lymphatics, where they present the viruses to the CD4+ T cells in the process of infecting them and spreading the infection [134]. Therefore, males who undergo circumcision have up to 60% reduced chances of contracting HIV because of the loss of the foreskin, which significantly reduces both CD4+ T cells and Langerhans cells [135][136][137]. It is a well-established fact that females generally mount stronger immune responses to both self and non-self-antigens, including viral infection: therefore, females are more prone to autoimmune disease than males [138]. While the mechanisms behind the sex differences are not fully understood, several genetic and physiological mechanisms are thought to be responsible for the stronger immune responses mounted by females compared to males in response to HIV infection and for how this may influence disease progression [139]. The first mechanism that can explain the stronger immune responses in females is the presence of X chromosome-linked genes that contribute to the higher expression of TLRs in females [140]. TLR7, which is higher in females, is located on the X chromosome, and females have two copies of the X chromosome, as opposed to males, who only have one [141]. The differences in immune response between sexes could also be attributed to sex hormones. Sex hormones have been shown to regulate the expression of TLRs, particularly TLR7 and TLR8, which recognize single-stranded RNA viruses such as HIV; this can lead to a stronger innate immune response against HIV and may help to control viral replication [142][143][144]. Estrogen has also been shown to stimulate immune response by increasing the number and activity of immune cells such as T cells and B cells, and it achieves this by stimulating the secretion of cytokines from a variety of cells, including immune cells such as monocytes, macrophages, dendritic cells and T cells [145][146][147]. Estrogen modulates the production of cytokines such as interleukin-1 (IL-1), interleukin-6 (IL-6), TNF-alpha and IFN-γ, among others [148][149][150][151][152][153]. These cytokines are important in regulating immune responses and play a role in inflammatory and autoimmune diseases [154][155][156][157]. Estrogen stimulates cytokine release by binding to the Estrogen receptors (ERs), ER-alpha (ERα) and ER-beta (ERβ), which are expressed on the surface of immune cells [158]. Estrogen can also bind to membrane-associated estrogen receptors (mERs), such as G protein-coupled receptor 30 (GPR30), which activate intracellular signaling cascades, such as the mitogen-activated protein kinase (MAPK) pathway [159][160][161]. These signaling pathways regulate cytokine production by immune cells [162][163]. The MAPK pathway can activate the c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38 MAPK sub-pathways, which regulate different aspects of immune cell activation [163].References
- German Advisory Committee Blood. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemotherapy 2016, 43, 203–222.
- Moss, J.A. HIV/AIDS Review. Radiol. Technol. 2013, 84, 247–267.
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. HIV Epidemiology. The Early Spread and Epidemic Ignition of HIV-1 in Human Populations. Science 2014, 346, 56–61.
- Rife, B.; Salemi, M. On the Early Dynamics and Spread of HIV-1. Trends Microbiol. 2015, 23, 3–4.
- Visseaux, B.; Le Hingrat, Q.; Damond, F.; Charpentier, C.; Descamps, D. . Virol. Montrouge Fr. 2019, 23, 277–291.
- Kalinichenko, S.; Komkov, D.; Mazurov, D. HIV-1 and HTLV-1 Transmission Modes: Mechanisms and Importance for Virus Spread. Viruses 2022, 14, 152.
- Visseaux, B.; Damond, F.; Matheron, S.; Descamps, D.; Charpentier, C. Hiv-2 Molecular Epidemiology. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 233–240.
- Boswell, M.T.; Rowland-Jones, S.L. Delayed Disease Progression in HIV-2: The Importance of TRIM5α and the Retroviral Capsid. Clin. Exp. Immunol. 2019, 196, 305–317.
- Vidya Vijayan, K.K.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front. Immunol. 2017, 8, 580.
- Joseph, S.B.; Arrildt, K.T.; Sturdevant, C.B.; Swanstrom, R. HIV-1 Target Cells in the CNS. J. Neurovirol. 2015, 21, 276–289.
- Woodham, A.W.; Skeate, J.G.; Sanna, A.M.; Taylor, J.R.; Da Silva, D.M.; Cannon, P.M.; Kast, W.M. Human Immunodeficiency Virus Immune Cell Receptors, Coreceptors, and Cofactors: Implications for Prevention and Treatment. AIDS Patient Care STDs 2016, 30, 291–306.
- Fenwick, C.; Joo, V.; Jacquier, P.; Noto, A.; Banga, R.; Perreau, M.; Pantaleo, G. T-Cell Exhaustion in HIV Infection. Immunol. Rev. 2019, 292, 149–163.
- Ma, T.; Luo, X.; George, A.F.; Mukherjee, G.; Sen, N.; Spitzer, T.L.; Giudice, L.C.; Greene, W.C.; Roan, N.R. HIV Efficiently Infects T Cells from the Endometrium and Remodels Them to Promote Systemic Viral Spread. eLife 2020, 9, e55487.
- Renault, C.; Veyrenche, N.; Mennechet, F.; Bedin, A.-S.; Routy, J.-P.; Van de Perre, P.; Reynes, J.; Tuaillon, E. Th17 CD4+ T-Cell as a Preferential Target for HIV Reservoirs. Front. Immunol. 2022, 13, 822576.
- Ng’eno, B.N.; Kellogg, T.A.; Kim, A.A.; Mwangi, A.; Mwangi, M.; Wamicwe, J.; Rutherford, G.W. Modes of HIV Transmission among Adolescents and Young Adults Aged 10-24 Years in Kenya. Int. J. STD AIDS 2018, 29, 800–805.
- del Rio, C. The Global HIV Epidemic: What the Pathologist Needs to Know. Semin. Diagn. Pathol. 2017, 34, 314–317.
- Kassa, G.M. Mother-to-Child Transmission of HIV Infection and Its Associated Factors in Ethiopia: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2018, 18, 216.
- Osório, D.; Munyangaju, I.; Nacarapa, E.; Muhiwa, A.; Nhangave, A.V.; Ramos, J.M. Mother-to-Child Transmission of HIV Infection and Its Associated Factors in the District of Bilene, Gaza Province-Mozambique. PLoS ONE 2021, 16, e0260941.
- Morar, M.M.; Pitman, J.P.; McFarland, W.; Bloch, E.M. The Contribution of Unsafe Blood Transfusion to Human Immunodeficiency Virus Incidence in Sub-Saharan Africa: Reexamination of the 5% to 10% Convention. Transfusion 2016, 56, 3121–3132.
- Belov, A.; Yang, H.; Forshee, R.A.; Whitaker, B.I.; Eder, A.F.; Chancey, C.; Anderson, S.A. Modeling the Risk of HIV Transfusion Transmission. J. Acquir. Immune Defic. Syndr. 1999 2023, 92, 173–179.
- Ball, L.J.; Puka, K.; Speechley, M.; Wong, R.; Hallam, B.; Wiener, J.C.; Koivu, S.; Silverman, M.S. Sharing of Injection Drug Preparation Equipment Is Associated With HIV Infection: A Cross-Sectional Study. J. Acquir. Immune Defic. Syndr. 1999 2019, 81, e99–e103.
- Kumar, P.; Aridoss, S.; Mathiyazhakan, M.; Balasubramanian, G.; Jaganathasamy, N.; Natesan, M.; Padmapriya, V.M.; David, J.K.; Rajan, S.; Adhikary, R.; et al. Substance Use and Risk of HIV Infection among Men Who Have Sex with Men in India. Medicine 2020, 99, e21360.
- Scheim, A.I.; Nosova, E.; Knight, R.; Hayashi, K.; Kerr, T. HIV Incidence Among Men Who Have Sex with Men and Inject Drugs in a Canadian Setting. AIDS Behav. 2018, 22, 3957–3961.
- El-Bassel, N.; Shaw, S.A.; Dasgupta, A.; Strathdee, S.A. Drug Use as a Driver of HIV Risks: Re-Emerging and Emerging Issues. Curr. Opin. HIV AIDS 2014, 9, 150–155.
- Edeza, A.; Bazzi, A.; Salhaney, P.; Biancarelli, D.; Childs, E.; Mimiaga, M.J.; Drainoni, M.-L.; Biello, K. HIV Pre-Exposure Prophylaxis for People Who Inject Drugs: The Context of Co-Occurring Injection- and Sexual-Related HIV Risk in the U.S. Northeast. Subst. Use Misuse 2020, 55, 525–533.
- Bruxelle, J.-F.; Trattnig, N.; Mureithi, M.W.; Landais, E.; Pantophlet, R. HIV-1 Entry and Prospects for Protecting against Infection. Microorganisms 2021, 9, 228.
- Jewanraj, J.; Ngcapu, S.; Liebenberg, L.J.P. Semen: A Modulator of Female Genital Tract Inflammation and a Vector for HIV-1 Transmission. Am. J. Reprod. Immunol. 2021, 86, e13478.
- Cavarelli, M.; Le Grand, R. The Importance of Semen Leukocytes in HIV-1 Transmission and the Development of Prevention Strategies. Hum. Vaccines Immunother. 2020, 16, 2018–2032.
- Kaul, R.; Liu, C.M.; Park, D.E.; Galiwango, R.M.; Tobian, A.A.R.; Prodger, J.L. The Penis, the Vagina and HIV Risk: Key Differences (Aside from the Obvious). Viruses 2022, 14, 1164.
- Jones, J.; Sullivan, P.S.; Curran, J.W. Progress in the HIV Epidemic: Identifying Goals and Measuring Success. PLoS Med. 2019, 16, e1002729.
- Shcherbatova, O.; Grebennikov, D.; Sazonov, I.; Meyerhans, A.; Bocharov, G. Modeling of the HIV-1 Life Cycle in Productively Infected Cells to Predict Novel Therapeutic Targets. Pathogens 2020, 9, 255.
- Nelson, A.G.; Zhang, X.; Ganapathi, U.; Szekely, Z.; Flexner, C.W.; Owen, A.; Sinko, P.J. Drug Delivery Strategies and Systems for HIV/AIDS Pre-Exposure Prophylaxis and Treatment. J. Control. Release 2015, 219, 669–680.
- Sankaranantham, M. HIV—Is a Cure Possible? Indian J. Sex. Transm. Dis. AIDS 2019, 40, 1–5.
- Sakuragi, J. Morphogenesis of the Infectious HIV-1 Virion. Front. Microbiol. 2011, 2, 242.
- McGettigan, J.P.; Naper, K.; Orenstein, J.; Koser, M.; McKenna, P.M.; Schnell, M.J. Functional Human Immunodeficiency Virus Type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and Env Expressed from a Single Rhabdovirus-Based Vaccine Vector Genome. J. Virol. 2003, 77, 10889–10899.
- Hill, M.; Tachedjian, G.; Mak, J. The Packaging and Maturation of the HIV-1 Pol Proteins. Curr. HIV Res. 2005, 3, 73–85.
- Staudt, R.P.; Alvarado, J.J.; Emert-Sedlak, L.A.; Shi, H.; Shu, S.T.; Wales, T.E.; Engen, J.R.; Smithgall, T.E. Structure, Function, and Inhibitor Targeting of HIV-1 Nef-Effector Kinase Complexes. J. Biol. Chem. 2020, 295, 15158–15171.
- NIH HIV Replication Cycle|NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/diseases-conditions/hiv-replication-cycle (accessed on 23 February 2023).
- Greta Hughson HIV Lifecycle. Available online: https://www.aidsmap.com/about-hiv/hiv-lifecycle (accessed on 3 March 2023).
- Xiao, T.; Cai, Y.; Chen, B. HIV-1 Entry and Membrane Fusion Inhibitors. Viruses 2021, 13, 735.
- Wilen, C.B.; Tilton, J.C.; Doms, W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866.
- Negi, G.; Sharma, A.; Dey, M.; Dhanawat, G.; Parveen, N. Membrane Attachment and Fusion of HIV-1, Influenza A, and SARS-CoV-2: Resolving the Mechanisms with Biophysical Methods. Biophys. Rev. 2022, 14, 1109–1140.
- Doms, R.W.; Moore, J.P. HIV-1 Membrane Fusion. J. Cell. Biol. 2000, 151, f9–f14.
- Sapp, N.; Burge, N.; Cox, K.; Prakash, P.; Balasubramaniam, M.; Thapa, S.; Christensen, D.; Li, M.; Linderberger, J.; Kvaratskhelia, M.; et al. HIV-1 Preintegration Complex Preferentially Integrates the Viral DNA into Nucleosomes Containing Trimethylated Histone 3-Lysine 36 Modification and Flanking Linker DNA. J. Virol. 2022, 96, e0101122.
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94, e00135-20.
- Rozina, A.; Anisenko, A.; Kikhai, T.; Silkina, M.; Gottikh, M. Complex Relationships between HIV-1 Integrase and Its Cellular Partners. Int. J. Mol. Sci. 2022, 23, 12341.
- Elliott, J.L.; Kutluay, S.B. Going beyond Integration: The Emerging Role of HIV-1 Integrase in Virion Morphogenesis. Viruses 2020, 12, 1005.
- PubChem HIV Life Cycle. Available online: https://pubchem.ncbi.nlm.nih.gov/pathway/Reactome:R-HSA-162587 (accessed on 3 March 2023).
- Goulder, P.J.R.; Watkins, D.I. Impact of MHC Class I Diversity on Immune Control of Immunodeficiency Virus Replication. Nat. Rev. Immunol. 2008, 8, 619–630.
- Swigut, T.; Alexander, L.; Morgan, J.; Lifson, J.; Mansfield, K.G.; Lang, S.; Johnson, R.P.; Skowronski, J.; Desrosiers, R. Impact of Nef-Mediated Downregulation of Major Histocompatibility Complex Class I on Immune Response to Simian Immunodeficiency Virus. J. Virol. 2004, 78, 13335–13344.
- Kerkau, T.; Bacik, I.; Bennink, J.R.; Yewdell, J.W.; Hünig, T.; Schimpl, A.; Schubert, U. The Human Immunodeficiency Virus Type 1 (HIV-1) Vpu Protein Interferes with an Early Step in the Biosynthesis of Major Histocompatibility Complex (MHC) Class I Molecules. J. Exp. Med. 1997, 185, 1295–1306.
- Koyanagi, N.; Kawaguchi, Y. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses 2020, 12, 1354.
- Stumptner-Cuvelette, P.; Morchoisne, S.; Dugast, M.; Le Gall, S.; Raposo, G.; Schwartz, O.; Benaroch, P. HIV-1 Nef Impairs MHC Class II Antigen Presentation and Surface Expression. Proc. Natl. Acad. Sci. USA 2001, 98, 12144–12149.
- Wonderlich, E.R.; Leonard, J.A.; Collins, K.L. HIV Immune Evasion: Disruption of Antigen Presentation by the HIV Nef Protein. Adv. Virus Res. 2011, 80, 103–127.
- Roeth, J.F.; Collins, K.L. Human Immunodeficiency Virus Type 1 Nef: Adapting to Intracellular Trafficking Pathways. Microbiol. Mol. Biol. Rev. 2006, 70, 548–563.
- Blagoveshchenskaya, A.D.; Thomas, L.; Feliciangeli, S.F.; Hung, C.-H.; Thomas, G. HIV-1 Nef Downregulates MHC-I by a PACS-1- and PI3K-Regulated ARF6 Endocytic Pathway. Cell 2002, 111, 853–866.
- Khan, N.; Geiger, J.D. Role of Viral Protein U (Vpu) in HIV-1 Infection and Pathogenesis. Viruses 2021, 13, 1466.
- Dubé, M.; Bego, M.G.; Paquay, C.; Cohen, É.A. Modulation of HIV-1-Host Interaction: Role of the Vpu Accessory Protein. Retrovirology 2010, 7, 114.
- Guha, D.; Ayyavoo, V. Innate Immune Evasion Strategies by Human Immunodeficiency Virus Type 1. ISRN AIDS 2013, 2013, 954806.
- Carrington, M.; Alter, G. Innate Immune Control of HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007070.
- Tomescu, C.; Law, W.K.; Kedes, D.H. Surface Downregulation of Major Histocompatibility Complex Class I, PE-CAM, and ICAM-1 Following De Novo Infection of Endothelial Cells with Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2003, 77, 9669–9684.
- Dirk, B.S.; Pawlak, E.N.; Johnson, A.L.; Van Nynatten, L.R.; Jacob, R.A.; Heit, B.; Dikeakos, J.D. HIV-1 Nef Sequesters MHC-I Intracellularly by Targeting Early Stages of Endocytosis and Recycling. Sci. Rep. 2016, 6, 37021.
- Mielke, D.; Bandawe, G.; Zheng, J.; Jones, J.; Abrahams, M.-R.; Bekker, V.; Ochsenbauer, C.; Garrett, N.; Abdool Karim, S.; Moore, P.L.; et al. ADCC-Mediating Non-Neutralizing Antibodies Can Exert Immune Pressure in Early HIV-1 Infection. PLoS Pathog. 2021, 17, e1010046.
- Martín, J.; LaBranche, C.C.; González-Scarano, F. Differential CD4/CCR5 Utilization, Gp120 Conformation, and Neutralization Sensitivity between Envelopes from a Microglia-Adapted Human Immunodeficiency Virus Type 1 and Its Parental Isolate. J. Virol. 2001, 75, 3568–3580.
- Raja, A.; Venturi, M.; Kwong, P.; Sodroski, J. CD4 Binding Site Antibodies Inhibit Human Immunodeficiency Virus Gp120 Envelope Glycoprotein Interaction with CCR5. J. Virol. 2003, 77, 713–718.
- Sachdeva, M.; Fischl, M.A.; Pahwa, R.; Sachdeva, N.; Pahwa, S. Immune Exhaustion Occurs Concomitantly with Immune Activation and Decrease in Regulatory T Cells in Viremic Chronically HIV-1 Infected Patients. J. Acquir. Immune Defic. Syndr. 1999 2010, 54, 447–454.
- Balasubramaniam, M.; Pandhare, J.; Dash, C. Immune Control of HIV. J. Life Sci. Westlake Village Calif. 2019, 1, 4–37.
- Walker, B.; McMichael, A. The T-Cell Response to HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007054.
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-Associated Lymphocyte Apoptosis: 2010. Cell. Death Dis. 2010, 1, e99.
- Oyaizu, N.; McCloskey, T.; Than, S.; Hu, R.; Kalyanaraman, V.; Pahwa, S. Cross-Linking of CD4 Molecules Upregulates Fas Antigen Expression in Lymphocytes by Inducing Interferon-Gamma and Tumor Necrosis Factor- Alpha Secretion. Blood 1994, 84, 2622–2631.
- Chandrasekar, A.P.; Cummins, N.W.; Badley, A.D. The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence. Clin. Microbiol. Rev. 2019, 33, e00107-19.
- Xu, H.; Wang, X.; Veazey, R.S. Mucosal Immunology of HIV Infection. Immunol. Rev. 2013, 254, 10–33.
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The Immune Response during Acute HIV-1 Infection: Clues for Vaccine Development. Nat. Rev. Immunol. 2010, 10, 11–23.
- Batorsky, R.; Sergeev, R.A.; Rouzine, I.M. The Route of HIV Escape from Immune Response Targeting Multiple Sites Is Determined by the Cost-Benefit Tradeoff of Escape Mutations. PLoS Comput. Biol. 2014, 10, e1003878.
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251.
- Abram, M.E.; Ferris, A.L.; Das, K. Mutations in HIV-1 Reverse Transcriptase Affect the Errors Made in a Single Cycle of Viral Replication. J. Virol. 2014, 88, 7589–7601.
- Burton, D.R.; Mascola, J.R. Antibody Responses to Envelope Glycoproteins in HIV-1 Infection. Nat. Immunol. 2015, 16, 571–576.
- Prabakaran, P.; Dimitrov, A.S.; Fouts, T.R.; Dimitrov, D.S. Structure and Function of the HIV Envelope Glycoprotein as Entry Mediator, Vaccine Immunogen, and Target for Inhibitors. Adv. Pharmacol. San. Diego Calif. 2007, 55, 33–97.
- Swiecki, M.; Colonna, M. Type I Interferons: Diversity of Sources, Production Pathways and Effects on Immune Responses. Curr. Opin. Virol. 2011, 1, 463–475.
- Marziali, F.; Delpeuch, M.; Kumar, A.; Appourchaux, R.; Dufloo, J.; Tartour, K.; Etienne, L.; Cimarelli, A. Functional Heterogeneity of Mammalian IFITM Proteins against HIV-1. J. Virol. 2021, 95, e0043921.
- Ali, S.; Mann-Nüttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778.
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49.
- Hermant, P.; Michiels, T. Interferon-λ in the Context of Viral Infections: Production, Response and Therapeutic Implications. J. Innate Immun. 2014, 6, 563–574.
- Lackner, A.A.; Lederman, M.M.; Rodriguez, B. HIV Pathogenesis: The Host. Cold Spring Harb. Perspect. Med. 2012, 2, a007005.
- Azimi, F.C.; Lee, J.E. Structural Perspectives on HIV-1 Vif and APOBEC3 Restriction Factor Interactions. Protein Sci. Publ. Protein Soc. 2020, 29, 391–406.
- Kung, W.-W.; Ramachandran, S.; Makukhin, N.; Bruno, E.; Ciulli, A. Structural Insights into Substrate Recognition by the SOCS2 E3 Ubiquitin Ligase. Nat. Commun. 2019, 10, 2534.
- Stanley, B.J.; Ehrlich, E.S.; Short, L.; Yu, Y.; Xiao, Z.; Yu, X.-F.; Xiong, Y. Structural Insight into the Human Immunodeficiency Virus Vif SOCS Box and Its Role in Human E3 Ubiquitin Ligase Assembly. J. Virol. 2008, 82, 8656.
- Lata, S.; Mishra, R.; Banerjea, A.C. Proteasomal Degradation Machinery: Favorite Target of HIV-1 Proteins. Front. Microbiol. 2018, 9, 2738.
- Du, Y.; Hu, Z.; Luo, Y.; Wang, H.Y.; Yu, X.; Wang, R.-F. Function and Regulation of CGAS-STING Signaling in Infectious Diseases. Front. Immunol. 2023, 14, 1130423.
- Sivro, A.; Su, R.-C.; Plummer, F.A.; Ball, T.B. Interferon Responses in HIV Infection: From Protection to Disease. AIDS Rev. 2014, 16, 43–51.
- Ananworanich, J.; Sacdalan, C.P.; Pinyakorn, S.; Chomont, N.; Souza, M.; Luekasemsuk, T.; Schuetz, A.; Krebs, S.J.; Dewar, R.; Jagodzinski, L.; et al. Virological and Immunological Characteristics of HIV-Infected Individuals at the Earliest Stage of Infection. J. Virus Erad. 2016, 2, 43–48.
- Robb, M.L.; Eller, L.A.; Kibuuka, H.; Rono, K.; Maganga, L.; Nitayaphan, S.; Kroon, E.; Sawe, F.K.; Sinei, S.; Sriplienchan, S.; et al. Prospective Study of Acute HIV-1 Infection in Adults in East Africa and Thailand. N. Engl. J. Med. 2016, 374, 2120–2130.
- Martín-Moreno, A.; Muñoz-Fernández, M.A. Dendritic Cells, the Double Agent in the War Against HIV-1. Front. Immunol. 2019, 10, 2485.
- Lee, H.-C.; Chathuranga, K.; Lee, J.-S. Intracellular Sensing of Viral Genomes and Viral Evasion. Exp. Mol. Med. 2019, 51, 1–13.
- Shi, Y.; Su, J.; Chen, R.; Wei, W.; Yuan, Z.; Chen, X.; Wang, X.; Liang, H.; Ye, L.; Jiang, J. The Role of Innate Immunity in Natural Elite Controllers of HIV-1 Infection. Front. Immunol. 2022, 13, 780922.
- Radoshevich, L.; Dussurget, O. Cytosolic Innate Immune Sensing and Signaling upon Infection. Front. Microbiol. 2016, 7, 313.
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869.
- Kell, A.M.; Gale, M. RIG-I in RNA Virus Recognition. Virology 2015, 479–480, 110–121.
- Abbas, F.; Cenac, C.; Youness, A.; Azar, P.; Delobel, P.; Guéry, J.-C. HIV-1 Infection Enhances Innate Function and TLR7 Expression in Female Plasmacytoid Dendritic Cells. Life Sci. Alliance 2022, 5, e202201452.
- Scagnolari, C.; Antonelli, G. Type I Interferon and HIV: Subtle Balance between Antiviral Activity, Immunopathogenesis and the Microbiome. Cytokine Growth Factor. Rev. 2018, 40, 19.
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291.
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379.
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384.
- Jang, J.-H.; Shin, H.W.; Lee, J.M.; Lee, H.-W.; Kim, E.-C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediat. Inflamm. 2015, 2015, 794143.
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273.
- Schoenemeyer, A.; Barnes, B.J.; Mancl, M.E.; Latz, E.; Goutagny, N.; Pitha, P.M.; Fitzgerald, K.A.; Golenbock, D.T. The Interferon Regulatory Factor, IRF5, Is a Central Mediator of Toll-like Receptor 7 Signaling. J. Biol. Chem. 2005, 280, 17005–17012.
- Kwa, M.Q.; Nguyen, T.; Huynh, J.; Ramnath, D.; De Nardo, D.; Lam, P.Y.; Reynolds, E.C.; Hamilton, J.A.; Sweet, M.J.; Scholz, G.M. Interferon Regulatory Factor 6 Differentially Regulates Toll-like Receptor 2-Dependent Chemokine Gene Expression in Epithelial Cells. J. Biol. Chem. 2014, 289, 19758–19768.
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic Cells in Progression and Pathology of HIV Infection. Trends Immunol. 2014, 35, 114–122.
- Chathuranga, K.; Weerawardhana, A.; Dodantenna, N.; Lee, J.-S. Regulation of Antiviral Innate Immune Signaling and Viral Evasion Following Viral Genome Sensing. Exp. Mol. Med. 2021, 53, 1647–1668.
- Collin, M.; Bigley, V. Human Dendritic Cell Subsets: An Update. Immunology 2018, 154, 3–20.
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92.
- Parihar, A.; Eubank, T.D.; Doseff, A.I. Monocytes and Macrophages Regulate Immunity through Dynamic Networks of Survival and Cell Death. J. Innate Immun. 2010, 2, 204–215.
- Mass, E.; Nimmerjahn, F.; Kierdorf, K.; Schlitzer, A. Tissue-Specific Macrophages: How They Develop and Choreograph Tissue Biology. Nat. Rev. Immunol. 2023, 1–17.
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90.
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804.
- Bajgar, A.; Krejčová, G. On the Origin of the Functional Versatility of Macrophages. Front. Physiol. 2023, 14, 246.
- Koppensteiner, H.; Brack-Werner, R.; Schindler, M. Macrophages and Their Relevance in Human Immunodeficiency Virus Type I Infection. Retrovirology 2012, 9, 82.
- Wu, L. Biology of HIV Mucosal Transmission. Curr. Opin. HIV AIDS 2008, 3, 534–540.
- Shen, R.; Richter, H.E.; Smith, P.D. Interactions between HIV-1 and Mucosal Cells in the Female Reproductive Tract. Am. J. Reprod. Immunol. 2014, 71, 608–617.
- Teer, E.; Joseph, D.E.; Glashoff, R.H.; Faadiel Essop, M. Monocyte/Macrophage-Mediated Innate Immunity in HIV-1 Infection: From Early Response to Late Dysregulation and Links to Cardiovascular Diseases Onset. Virol. Sin. 2021, 36, 565–576.
- Bertram, K.M.; Tong, O.; Royle, C.; Turville, S.G.; Nasr, N.; Cunningham, A.L.; Harman, A.N. Manipulation of Mononuclear Phagocytes by HIV: Implications for Early Transmission Events. Front. Immunol. 2019, 10, 2263.
- Okoye, A.A.; Picker, L.J. CD4+ T Cell Depletion in HIV Infection: Mechanisms of Immunological Failure. Immunol. Rev. 2013, 254, 54–64.
- Abbas, W.; Herbein, G. T-Cell Signaling in HIV-1 Infection. Open Virol. J. 2013, 7, 57–71.
- Courtney, A.H.; Lo, W.-L.; Weiss, A. TCR signaling: Mechanisms of initiation and propagation. Trends Biochem. Sci. 2018, 43, 108–123.
- Pino, S.C.; O’Sullivan-Murphy, B.; Lidstone, E.A.; Thornley, T.B.; Jurczyk, A.; Urano, F.; Greiner, D.L.; Mordes, J.P.; Rossini, A.A.; Bortell, R. Protein Kinase C Signaling during T Cell Activation Induces the Endoplasmic Reticulum Stress Response. Cell. Stress. Chaperones 2008, 13, 421–434.
- Perdomo-Celis, F.; Taborda, N.A.; Rugeles, M.T. CD8+ T-Cell Response to HIV Infection in the Era of Antiretroviral Therapy. Front. Immunol. 2019, 10, 1896.
- McBrien, J.B.; Kumar, N.A.; Silvestri, G. Mechanisms of CD8+ T Cell-Mediated Suppression of HIV/SIV Replication. Eur. J. Immunol. 2018, 48, 898–914.
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C.M.L. Cytotoxic CD4 T Cells—Friend or Foe during Viral Infection? Front. Immunol. 2017, 8, 19.
- Ziegler, S.; Altfeld, M. Sex Differences in HIV-1-Mediated Immunopathology. Curr. Opin. HIV AIDS 2016, 11, 209.
- Santinelli, L.; Ceccarelli, G.; Borrazzo, C.; Innocenti, G.P.; Frasca, F.; Cavallari, E.N.; Celani, L.; Nonne, C.; Mastroianni, C.M.; d’Ettorre, G. Sex-Related Differences in Markers of Immune Activation in Virologically Suppressed HIV-Infected Patients. Biol. Sex. Differ. 2020, 11, 23.
- Anastos, K.; Gange, S.J.; Lau, B.; Weiser, B.; Detels, R.; Giorgi, J.V.; Margolick, J.B.; Cohen, M.; Phair, J.; Melnick, S.; et al. Association of Race and Gender with HIV-1 RNA Levels and Immunologic Progression. J. Acquir. Immune Defic. Syndr. 2000, 24, 218–226.
- Portales, P.; Clot, J.; Corbeau, P. Sex Differences in HIV-1 Viral Load Due to Sex Difference in CCR5 Expression. Ann. Intern. Med. 2001, 134, 81–82.
- Prodger, J.L.; Gray, R.; Kigozi, G.; Nalugoda, F.; Galiwango, R.; Hirbod, T.; Wawer, M.; Hofer, S.O.P.; Sewankambo, N.; Serwadda, D.; et al. Foreskin T Cell Subsets Differ Substantially from Blood with Respect to HIV Co-Receptor Expression, Inflammatory Profile and Memory Status. Mucosal Immunol. 2012, 5, 121–128.
- Ballweber, L.; Robinson, B.; Kreger, A.; Fialkow, M.; Lentz, G.; McElrath, M.J.; Hladik, F. Vaginal Langerhans Cells Nonproductively Transporting HIV-1 Mediate Infection of T Cells. J. Virol. 2011, 85, 13443–13447.
- Rasheed, Z. Male Circumcision and Human Immunodeficiency Virus Infection: An Update on Randomized Controlled Trials and Molecular Evidences. Int. J. Health Sci. 2018, 12, 1–3.
- Prodger, J.L.; Kaul, R. The Biology of How Circumcision Reduces HIV Susceptibility: Broader Implications for the Prevention Field. AIDS Res. Ther. 2017, 14, 49.
- Weiss, H.A.; Dickson, K.E.; Agot, K.; Hankins, C.A. Male Circumcision for HIV Prevention: Current Research and Programmatic Issues. AIDS Lond. Engl. 2010, 24 (Suppl. 4), S61–S69.
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638.
- Moran, J.A.; Turner, S.R.; Marsden, M.D. Contribution of Sex Differences to HIV Immunology, Pathogenesis, and Cure Approaches. Front. Immunol. 2022, 13, 905773.
- Addo, M.M.; Altfeld, M. Sex-Based Differences in HIV Type 1 Pathogenesis. J. Infect. Dis. 2014, 209, S86–S92.
- Youness, A.; Miquel, C.-H.; Guéry, J.-C. Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 1114.
- Young, N.A.; Wu, L.-C.; Burd, C.J.; Friedman, A.K.; Kaffenberger, B.H.; Rajaram, M.V.S.; Schlesinger, L.S.; James, H.; Shupnik, M.A.; Jarjour, W.N. Estrogen Modulation of Endosome-Associated Toll-like Receptor 8: An IFNα-Independent Mechanism of Sex-Bias in Systemic Lupus Erythematosus. Clin. Immunol. 2014, 151, 66–77.
- Justina, V.D.; Giachini, F.R.; Sullivan, J.C.; Webb, R.C. Toll-Like Receptors Contribute to Sex Differences in Blood Pressure Regulation. J. Cardiovasc. Pharmacol. 2020, 76, 255–266.
- Zandieh, Z.; Amjadi, F.; Ashrafi, M.; Aflatoonian, A.; Fazeli, A.; Aflatoonian, R. The Effect of Estradiol and Progesterone on Toll Like Receptor Gene Expression in A Human Fallopian Tube Epithelial Cell Line. Cell. J. Yakhteh 2016, 17, 678–691.
- Harding, A.T.; Heaton, N.S. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers 2022, 14, 909.
- Alesci, A.; Nicosia, N.; Fumia, A.; Giorgianni, F.; Santini, A.; Cicero, N. Resveratrol and Immune Cells: A Link to Improve Human Health. Molecules 2022, 27, 424.
- Liao, Z.H.; Huang, T.; Xiao, J.W.; Gu, R.C.; Ouyang, J.; Wu, G.; Liao, H. Estrogen Signaling Effects on Muscle-Specific Immune Responses through Controlling the Recruitment and Function of Macrophages and T Cells. Skelet. Muscle 2019, 9, 20.
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295.
- Cai, Y.; Zhou, J.; Webb, D.C. Estrogen Stimulates Th2 Cytokine Production and Regulates the Compartmentalisation of Eosinophils during Allergen Challenge in a Mouse Model of Asthma. Int. Arch. Allergy Immunol. 2012, 158, 252–260.
- Joseph, K.; Tholanikunnel, B.G.; Kaplan, A.P. Cytokine and Estrogen Stimulation of Endothelial Cells Augments Activation of the Prekallikrein-High Molecular Weight Kininogen Complex: Implications for Hereditary Angioedema. J. Allergy Clin. Immunol. 2017, 140, 170–176.
- Shivers, K.-Y.; Amador, N.; Abrams, L.; Hunter, D.; Jenab, S.; Quiñones-Jenab, V. Estrogen Alters Baseline and Inflammatory-Induced Cytokine Levels Independent from Hypothalamic–Pituitary–Adrenal Axis Activity. Cytokine 2015, 72, 121–129.
- Cutolo, M.; Sulli, A.; Straub, R.H. Estrogen Metabolism and Autoimmunity. Autoimmun. Rev. 2012, 11, A460–A464.
- Javadian, A.; Salehi, E.; Bidad, K.; Sahraian, M.A.; Izad, M. Effect of Estrogen on Th1, Th2 and Th17 Cytokines Production by Proteolipid Protein and PHA Activated Peripheral Blood Mononuclear Cells Isolated from Multiple Sclerosis Patients. Arch. Med. Res. 2014, 45, 177–182.
- Gao, H.; Liu, L.; Zhao, Y.; Hara, H.; Chen, P.; Xu, J.; Tang, J.; Wei, L.; Li, Z.; Cooper, D.K.C.; et al. Human IL-6, IL-17, IL-1β, and TNF-α Differently Regulate the Expression of pro-Inflammatory Related Genes, Tissue Factor, and Swine Leukocyte Antigen Class I in Porcine Aortic Endothelial Cells. Xenotransplantation 2017, 24, e12291.
- Hagman, S.; Mäkinen, A.; Ylä-Outinen, L.; Huhtala, H.; Elovaara, I.; Narkilahti, S. Effects of Inflammatory Cytokines IFN-γ, TNF-α and IL-6 on the Viability and Functionality of Human Pluripotent Stem Cell-Derived Neural Cells. J. Neuroimmunol. 2019, 331, 36–45.
- Theofilopoulos, A.N.; Koundouris, S.; Kono, D.H.; Lawson, B.R. The Role of IFN-Gamma in Systemic Lupus Erythematosus: A Challenge to the Th1/Th2 Paradigm in Autoimmunity. Arthritis Res. Ther. 2001, 3, 136.
- Chaouali, M.; Azaiez, M.B.; Tezeghdenti, A.; Yacoubi-Oueslati, B.; Ghazouani, E.; Kochkar, R. High Levels of Proinflammatory Cytokines IL-6, IL-8, TNF-A, IL-23, and IFN-γ in Tunisian Patients with Type 1 Autoimmune Hepatitis. Eur. Cytokine Netw. 2020, 31, 94–103.
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen Receptors Alpha (ERα) and Beta (ERβ): Subtype-Selective Ligands and Clinical Potential. Steroids 2014, 0, 13–29.
- Atanaskova, N.; Keshamouni, V.G.; Krueger, J.S.; Schwartz, J.A.; Miller, F.; Reddy, K.B. MAP Kinase/Estrogen Receptor Cross-Talk Enhances Estrogen-Mediated Signaling and Tumor Growth but Does Not Confer Tamoxifen Resistance. Oncogene 2002, 21, 4000–4008.
- Liu, A.; Zhang, D.; Yang, X.; Song, Y. Estrogen Receptor Alpha Activates MAPK Signaling Pathway to Promote the Development of Endometrial Cancer. J. Cell. Biochem. 2019, 120, 17593–17601.
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen Action via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and CAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol. Endocrinol. 2002, 16, 70–84.
- Petrova, T.; Pesic, J.; Pardali, K.; Gaestel, M.; Arthur, J.S.C. P38 MAPK Signalling Regulates Cytokine Production in IL-33 Stimulated Type 2 Innate Lymphoid Cells. Sci. Rep. 2020, 10, 3479.
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254.