Being immune privileged, the central nervous system (CNS) is populated by unique parenchymal and non-parenchymal tissue-resident macrophages, namely, microglia and border-associated macrophages (BAMs), respectively. BAMs are found in the choroid plexus, meningeal and perivascular spaces, playing critical roles in maintaining CNS homeostasis while being phenotypically and functionally distinct from microglial cells. Although the ontogeny of microglia has been largely determined, BAMs need comparable scrutiny as they have been recently discovered and have not been thoroughly explored. Shedding light on the molecular cues and drivers orchestrating BAM generation is essential for delineating their cellular identity. BAMs are receiving more attention since they are gradually incorporated into neurodegenerative and neuroinflammatory disease evaluations. Understanding the ontogeny of BAMs and their involvement in CNS diseases paves the way for targeted therapeutic strategies and precision medicine.
- CNS border-associated macrophages
- tissue-resident macrophages
- origin
- yolk sac
- molecular cues
- development
- disease
1. Introduction
2. Origin of BAMs during Embryogenesis and Adulthood
The BAM embryonic origin was first investigated in rodents using bone marrow chimeras and whole-body irradiation, proposing that BAMs are bone marrow-derived [37,38][37][38]. In 2016, Goldmann et al., performing fate mapping analysis, observed that BAMs originate from the mouse yolk sac’s early erythro-myeloid progenitors (EMPs) during embryogenesis [8]. A tamoxifen-inducible Runx1CreERR26YFPfate-mapping mouse model confirmed that BAMs originate from early EMPs in the yolk sac, which gave rise to two different macrophage populations, namely, CD206+ (BAM progenitors) and CD206− (microglial progenitors) without the contribution of fetal liver or definitive hematopoiesis [20]. Interestingly, the mannose receptor C-type 1 (MRC1 or CD206) is a unique marker for BAMs [8,12,14][8][12][14]. The expression of Mrc1 is upregulated from E8.5 when the primitive macrophages, originating from EMPs, prepare to invade the embryonic tissues [39]. Recently, Masuda et al. investigated the progenitors of BAMs utilizing single-cell RNA sequencing and fate mapping analysis in the Mrc1CreERT2 mouse model. Although flow cytometry confirmed the presence of a CD206+ subpopulation within the A2 cells (CD45+ c-kit− CX3CR1+ cells), meningeal macrophages and microglia were found to originate from common CD206+ A2 progenitors in contrast with previous results [20,40][20][40]. The pvΜΦ were generated postnatally from sdΜΦ, requiring integrin-signaling and vascular smooth muscle cells (VSMCs) [40]. Regarding the repopulation pattern of BAMs in adulthood, there is a great heterogeneity between BAM clusters; specifically, the sdΜΦ, pvMΦ and cpepiΜΦ exhibit similar longevity with microglial cells as being self-maintained in the CNS independently from blood monocytes’ contribution [8,14][8][14]. The cpepiΜΦ were solely derived from local SALL1+ macrophages [14]. In Ccr2-deficient mice, the number of cpMΦ decreased, revealing their replenishment from Ly6Chi monocytes and shorter turnover [8]. In accordance with these results, Van Hove et al., combining single-cell RNA sequencing with complementary approaches in mice, suggested that dmΜΦ and cpΜΦ were gradually replenished by bone marrow-derived monocytes [14]. As dura mater and choroid plexus stroma are more accessible brain regions than (i) subdural space, (ii) the apical surface of the choroid plexuses, and (iii) brain parenchyma, the tissue permeability may be considered a crucial factor for brain macrophage ontogeny. However, the ablation of BAMs through CSF1R blockade led to the replenishment of cpΜΦ and dmΜΦ via local expansion, indicating their self-renewal capacity, while sdΜΦ presented difficulties in their repopulation [14]. By utilizing the Cx3cr1CreER:R26tdTomato fate mapping system in an experimental autoimmune encephalomyelitis (EAE) mouse model, Jordão et al. proposed that BAMs remained stable and locally self-renewed in addition to the recruitment of bone marrow-derived progenitors [12]. In Cx3cr1gfpCcr2rfp bone marrow chimeric mice, CD169+ BAMs proliferated after ischemia, while a small proportion of BAMs was bone marrow-derived, populating the perivascular and ischemic regions [36]. Both in homeostasis and disease, skull and vertebrae bone marrow constitute a pool of myeloid cells that can invade non-parenchymal and parenchymal CNS regions, transforming into tissue-resident macrophages [41]. A fate-mapping analysis in a mouse model of Alzheimer’s disease (AD) revealed that BAMs are a stable cell population with an unaffected turnover rate and a minimal replenishment from bone marrow-derived cells during this neurodegenerative disease [42]. Summarizing, the origin of BAMs has been extensively studied in the last few years using new genetic tools, e.g., fate mapping analysis. It has been proposed that BAMs originate from early EMPs in the yolk sac during embryogenesis. Although specific BAMs are replenished by peripherally-derived monocytes postnatally, some remain solely derived from the local pool. BAMs have been shown to remain stable and locally self-renewed in both homeostasis and disease. Further investigation is needed to (i) confirm BAM origin, (ii) detect the precise embryonic progenitors of BAMs, especially of the dura mater and choroid plexus macrophages, (iii) determine the timing of each BAM subpopulation’s generation, and (iv) delineate their repopulation pattern.3. Molecular Drives Orchestrating BAM Development
The transcription factor PU.1 (or SFPI) could be essential for the BAM generation during embryonic development since research has showed that in mice with deletion of the Sfpi1 gene, pvMΦ, sdMΦ, and cpMΦ were ablated [8]. Progenitors of BAMs express the runt-related transcription factor 1 (RUNX1), which regulates the expression of PU.1 during embryogenesis [20,43][20][43]. The impairment of PU.1 factor in mice results in a reduced number of A1 (CD45+ c-kitlo CX3CR1− immature cells) and A2 (CD45+ c-kit− CX3CR1+ cells) progenitor cells of the yolk sac, from which both microglial cells and BAMs originate. In contrast, the lack of interferon regulatory factor 8 (IRF8) exclusively decreased the number of A2 cells [44]. Furthermore, the colony-stimulating factor 1 receptor (CSF1R) signaling could be essential for BAM development [5,11,14][5][11][14]. In a zebrafish model carrying the panther mutation, a loss-of-function mutation in the fms gene orthologue which encodes CSF1R, primitive macrophages of the yolk sac could not colonize the embryonic tissues [45]. After progenitors’ migration and invasion in the CNS, BAM generation is initiated (Figure 1). The BAMs may be developed independently of transforming growth factor beta receptor (TGF-βR) signaling. In Tgfbr2-deficient mice, no alteration in cell numbers of BAMs occurred, while transforming growth factor beta (TGF-β) is required for the generation of microglial cells [20,46][20][46]. Three main brain border regions are filled with BAMs, namely, meninges, choroid plexus, and perivascular spaces. The postnatal expansion of sdMΦ was influenced by IRF8 and MAFB [40]. Indeed, in Irf8-deficient mice, a reduction of sdMΦ was observed [8]. The lack of integrin subunit beta 1 (ITGB1) in mice resulted only in a minor change in the numbers of sdΜΦ [40]. Similarly, the absence of insulin-like growth factor 1 (IGF1R) induces transcriptomic changes in BAMs via its implication in RNA processing, growth, migration and intracellular signaling [47]. The MYB, BATF3, and NR4A1 transcription factors were not necessary for BAM development [8].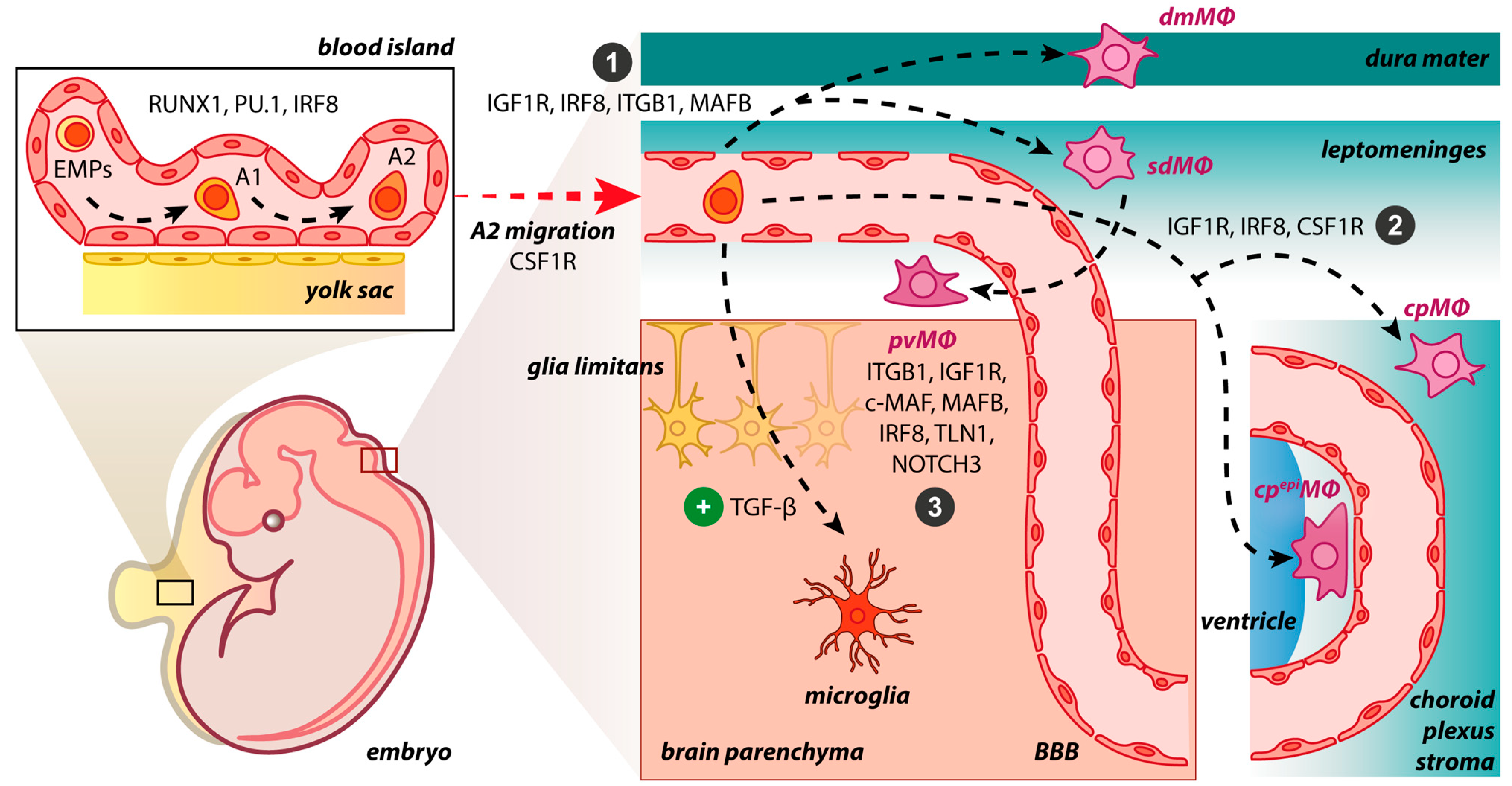
4. BAMs vs. Microglia
Although microglia and BAMs are immune-competent cells of the CNS with common progenitors, their different localization may contribute to variations in their biological roles. The microglial populations’ functions have been reviewed in detail [65,66,67][57][58][59]. Concisely, microglial cells are involved in developmental processes, including cell positioning, survival, myelinogenesis, synaptic patterning, and axonal dynamics [68][60]. In adult CNS, microglia, as the regulators of acute and chronic immune responses, are implicated in removing pathogens and noxious particles, scavenging cellular debris and synapses, protecting neural tissue, and mediating neurogenesis in CNS injury [24,66,69][24][58][61]. The unique localization of BAMs between brain parenchyma and peripheral tissues pinpoint their pivotal role in the immune surveillance of pathological antigens [16]. Their antigen-presenting capacity is attributed to MHC II molecules on some BAM surfaces [12,32,70,71][12][32][62][63]. Furthermore, the pvΜΦ and dmΜΦ mainly phagocytose intruding pathogens and any foreign molecule or substance that can be detected in the bloodstream and cerebrospinal fluid [72,72][64][64]. The pvΜΦ also appear to regulate the accessibility of brain parenchyma to circulating cells and molecules by increasing the contractility of regional vessels and capillaries or diminishing the BBB permeability [73,74,75,76][65][66][67][68]. Interestingly, the latest approaches demonstrate the involvement of BAMs in ensuring a well-balanced metabolic environment for neurons, especially in the course of systemic perturbations [77,78][69][70]. Data regarding morphology, motility, and molecular identity of microglia and BAMs are summarized in Table 1.| Cell Type |
Cell Type |
Morphology |
Morphology |
Motility |
Motility |
Cell-Specific Markers |
Cell-Specific Markers |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Microglia |
Microglia |
Ramified in homeostasis; Amoeboid in inflammation |
Ramified in homeostasis; Amoeboid in inflammation |
Cell bodies with limited-motility but highly dynamic processes in homeostasis; Highly phagocytic in inflammation |
Cell bodies with limited-motility but highly dynamic processes in homeostasis; Highly phagocytic in inflammation |
SIGLEC-H+, P2RY12 | , P2RY12+ | +, HEXB | , HEXB+, TMEM119+, ANXA3+, SALL1+ | +, TMEM119+, ANXA3+, SALL1+ |
SIGLEC-H+ |
| pvΜΦ |
pvΜΦ |
Slightly elongated cell bodies |
Slightly elongated cell bodies |
Non-motile cell bodies with extending and retracting projections through the blood vessel wall in homeostasis; Dendritic-like processes in inflammation |
Non-motile cell bodies with extending and retracting projections through the blood vessel wall in homeostasis; Dendritic-like processes in inflammation |
CD206+, CD38+, LYVE1+, CD36+, CD163+, CD169+ |
CD206+, CD38+, LYVE1+, CD36+, CD163+, CD169+ |
||||
| dmΜΦ |
dmΜΦ |
Elongated; Spindle-shaped cells; Few thick membrane projections; Dendriform |
Elongated; Spindle-shaped cells; Few thick membrane projections; Dendriform |
Limited motility and highly dynamic protrusions in homeostasis; Extending projections in inflammation |
Limited motility and highly dynamic protrusions in homeostasis; Extending projections in inflammation |
||||||
| sdΜΦ |
sdΜΦ |
Elongated; Amoeboid; Spindle-shaped cells; Few thick membrane projections |
Elongated; Amoeboid; Spindle-shaped cells; Few thick membrane projections |
Limited motility and highly dynamic protrusions in homeostasis; Extending projections in inflammation |
Limited motility and highly dynamic protrusions in homeostasis; Extending projections in inflammation |
||||||
| cpΜΦ |
cpΜΦ |
Star-like shape |
Star-like shape |
Unknown |
Unknown |
||||||
| cpepiΜΦ |
cpepiΜΦ |
Round; Bipolar; Stellate |
Round; Bipolar; Stellate |
Unknown |
Unknown |
5. BAMs in Neurological Diseases and Promising Therapies
The implication of BAMs in the pathogenesis of CNS diseases, especially in neurodegeneration and neuroinflammation, is a rapidly emerging field of research. Although the precise role of BAMs in diseases is not yet elucidated, recent studies have addressed their potential involvement in several pathological conditions such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and stroke. Further experimental studies are needed to delineate the exact pathophysiological mechanism through which methodical manipulation of BAMs can halt or even reverse the progression of the aforementioned debilitating CNS diseases.
5.1. BAMs in Alzheimer’s Disease
AD is a brain disorder constituting a common cause of dementia, characterized by permanent neurodegeneration in specific brain areas [86][71]. However, the pathophysiology of the disease is not yet fully understood. The accumulation of amyloid beta (Aβ) protein in the brain has been implicated in AD. This protein forms sticky plaques that may disrupt the interaction between brain cells, leading to inflammation and neuronal death [87][72]. Additionally, AD is characterized by the accumulation of the tau protein, which forms neurofibrillary tangles [88][73]. Patients with Alzheimer’s disease (AD) could be affected by cerebral amyloid angiopathy (CAA), which involves the pathologic deposition of Aβ within the leptomeningeal and cortical blood vessels [89][74]. The role of pvΜΦ in CAA progression has been investigated in a TgCRND8 mouse model of AD [90][75]. Hawkes and McLaurin demonstrated that the stimulation of the pvΜΦ turnover decreased cerebral CAA load. Interestingly, the clearance of CAA load was not attributed to microglia or astrocytes. These findings indicate the importance of pvΜΦ in CAA progression, suggesting that their activation could be a useful therapeutic approach for removing vascular amyloid [90][75]. In Tg2576 mice, the clodronate-mediated depletion of pvΜΦ reduced the production of reactive oxygen species, thereby reversing cerebrovascular dysfunction induced by Aβ. Experiments utilizing bone marrow chimeras revealed that pvΜΦ are the primary cell expressing CD36 and NOX2, which are molecular substrates for inducing cerebrovascular oxidative stress [91][76]. The pvΜΦ play a significant role in upregulating secreted phosphoprotein 1 (SPP1), with perivascular fibroblasts contributing to a lesser extent. SPP1 assists microglia in engulfing synapses and increases the expression of phagocytic markers such as complement C1q A chain (C1QA), granulin precursor (GRN), and cathepsin B (CTSB) in the presence of Aβ oligomers. The deletion of Spp1 in AD mouse models prevented synaptic loss [34]. Finally, the minor replenishment of CD206+ BAMs and their stable turnover in a mouse AD model should be highlighted, as potential manipulations of these cells could lead to modification of AD pathology [42].5.2. BAMs in Parkinson’s Disease
Parkinson’s disease (PD) is another common neurodegenerative disorder characterized by dopaminergic cell loss [92][77]. The accumulation of a-synuclein (α-SYN) is a distinct trait of degenerating dopaminergic neurons [93,94][78][79]. According to Guo et al., exosomes derived from microglia and CNS macrophages facilitated the transmission of α-SYN, leading to its aggregation in neurons and contributing to the development of PD [95][80]. Interestingly, BAMs may mediate the α-SYN related neuroinflammation by acting as antigen-presenting cells essential for initiating a CD4 T cell response [96][81]. The immune cell infiltration, recruitment, and antigen presentation were found to be greatly dependent on BAMs, framing their involvement in the pathogenesis of PD [96][81]. A JAK1/2 inhibitor, namely, AZD1480, has been considered a therapeutic option for PD by reducing α-SYN-related neuroinflammation via downregulation of the JAK/STAT pathway [97][82].5.3. BAMs in Multiple Sclerosis
Multiple sclerosis (MS) is a debilitating neurodegenerative disease with a rising global prevalence in recent years [98][83]. MS features encompass neuroinflammation, demyelination, and axonal loss within the CNS [99,100][84][85]. Several mechanisms have been proposed to be implicated in the pathophysiology of MS [101,102,103][86][87][88]. Nevertheless, the potential role of BAMs in MS has been only recently investigated [47,104][47][89]. BAMs, as a CNS macrophage population, could potentially be involved in the MS course through the CNS-targeted autoimmunity or neurodegeneration leading to a secondary autoimmune response [105][90]. BAMs are presented with different phenotypes regarding their roles in each stage of the MS [106][91]. Particularly, Locatelli et al. identified various markers of BAMs, utilizing immunofluorescent techniques in a MS mouse model, as the neuroinflammatory lesions shifted from expansion to gradual resolution [107][92]. In EAE, the most widely used animal model for studying MS aspects [108[93][94],109], antigen presentation and T cell reactivation were found to be regulated by both meningeal macrophages and microglia, revealing the involvement of BAMs in the disease [110,111][95][96]. The pvΜΦ and sdΜΦ were found to be modestly increased in the EAE mouse model, with sdΜΦ population expanding during disease onset, suggesting their implication in the initial acute phase of EAE. On the contrary, the sdΜΦ population decreased during the chronic phase of the disease and pvΜΦ proliferation remained unaltered [12]. The BAMs could also exert miscellaneous functions in MS via interleukin 9 (IL9) upregulation. Donninelli et al. found that MS patients had higher IL9 levels in the cerebrospinal fluid obtained from post-mortem samples. Through flow cytometry of snap-frozen tissue blocks from the same patients’ brains, higher expression of IL9 was also observed in macrophages [112][97]. Additionally, the disease-mediated peroxisome injury in BAMs, leading to demyelination and axonal loss, may be prevented through treatment with 4-Phenylbutyrate, which serves as a potential therapeutic approach for halting inflammatory demyelination and the progression of MS [113][98]. Lastly, foamy macrophages are formed in brain regions during MS; by targeting lipophagy, remyelination can be promoted as some BAM subtypes may be involved in the aforementioned process [114,115][99][100].5.4. BAMs in Other CNS Diseases
The BAMs have also been implicated in other CNS diseases, such as stroke. The study of Pedragosa et al. highlighted the major role of BAMs in different pathophysiological changes related to ischemic stroke, including the recruitment of granulocytes, increased expression of vascular endothelial growth factor (VEGF), and increased permeability of pial and cortical blood vessels [116][101]. The induction of ischemic stroke resulted in the proliferation and migration of CD163+ BAMs adopting a pro-inflammatory phenotype in the ischemic rat parenchyma. Although CD169+ perivascular macrophages were also observed to proliferate in response to ischemic stroke, they were replaced by infiltrating bone marrow-derived cells in mice. These findings were confirmed in a human model in which CD163+ cells were also accumulated in the ischemic region [36]. In the subarachnoid hemorrhage (SAH), sdΜΦ and pvΜΦ are involved in erythrocyte uptake affecting the outcome of hemorrhage. Specifically, their depletion led to the reduction of large arterioles’ inflammation and microthrombosis after SAH [117][102]. Ultimately, an induction of anti-inflammatory microglial/macrophage responses and subsequent neuroprotection could be achieved through the peripheral administration of interleukin 13 in cases of ischemic stroke [118][103].References
- Mastorakos, P.; McGavern, D. The Anatomy and Immunology of Vasculature in the Central Nervous System. Sci. Immunol. 2019, 4, eaav0492.
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242.
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790.
- Casano, A.M.; Peri, F. Microglia: Multitasking Specialists of the Brain. Dev. Cell 2015, 32, 469–477.
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6.
- Bechmann, I.; Kwidzinski, E.; Kovac, A.D.; Simbürger, E.; Horvath, T.; Gimsa, U.; Dirnagl, U.; Priller, J.; Nitsch, R. Turnover of Rat Brain Perivascular Cells. Exp. Neurol. 2001, 168, 242–249.
- Mato, M.; Ookawara, S.; Kurihara, K. Uptake of Exogenous Substances and Marked Infoldings of the Fluorescent Granular Pericyte in Cerebral Fine Vessels. Am. J. Anat. 1980, 157, 329–332.
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces. Nat. Immunol. 2016, 17, 797–805.
- McKinsey, G.L.; Lizama, C.O.; Keown-Lang, A.E.; Niu, A.; Santander, N.; Larpthaveesarp, A.; Chee, E.; Gonzalez, F.F.; Arnold, T.D. A New Genetic Strategy for Targeting Microglia in Development and Disease. eLife 2020, 9, e54590.
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; d’Errico, P.; Snaidero, N.; Costa Jordão, M.J.; Böttcher, C.; Kierdorf, K.; et al. Novel Hexb-Based Tools for Studying Microglia in the CNS. Nat. Immunol. 2020, 21, 802–815.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845.
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Locatelli, G.; Tai, Y.-H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; Groß, O.; et al. Single-Cell Profiling Identifies Myeloid Cell Subsets with Distinct Fates during Neuroinflammation. Science 2019, 363, eaat7554.
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91.
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A Single-Cell Atlas of Mouse Brain Macrophages Reveals Unique Transcriptional Identities Shaped by Ontogeny and Tissue Environment. Nat. Neurosci. 2019, 22, 1021–1035.
- Zelco, A.; Börjesson, V.; de Kanter, J.K.; Lebrero-Fernandez, C.; Lauschke, V.M.; Rocha-Ferreira, E.; Nilsson, G.; Nair, S.; Svedin, P.; Bemark, M.; et al. Single-Cell Atlas Reveals Meningeal Leukocyte Heterogeneity in the Developing Mouse Brain. Genes Dev. 2021, 35, 1190–1207.
- Mildenberger, W.; Stifter, S.A.; Greter, M. Diversity and Function of Brain-Associated Macrophages. Curr. Opin. Immunol. 2022, 76, 102181.
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The Mononuclear Phagocyte System: A New Classification of Macrophages, Monocytes, and Their Precursor Cells. Bull. World Health Organ. 1972, 46, 845–852.
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity 2015, 42, 665–678.
- Hickey, W.F.; Kimura, H. Perivascular Microglial Cells of the CNS Are Bone Marrow-Derived and Present Antigen in Vivo. Science 1988, 239, 290–292.
- Utz, S.G.; See, P.; Mildenberger, W.; Thion, M.S.; Silvin, A.; Lutz, M.; Ingelfinger, F.; Rayan, N.A.; Lelios, I.; Buttgereit, A.; et al. Early Fate Defines Microglia and Non-Parenchymal Brain Macrophage Development. Cell 2020, 181, 557–573.e18.
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449.
- Shemer, A.; Grozovski, J.; Tay, T.L.; Tao, J.; Volaski, A.; Süß, P.; Ardura-Fabregat, A.; Gross-Vered, M.; Kim, J.-S.; David, E.; et al. Engrafted Parenchymal Brain Macrophages Differ from Microglia in Transcriptome, Chromatin Landscape and Response to Challenge. Nat. Commun. 2018, 9, 5206.
- Stremmel, C.; Schuchert, R.; Wagner, F.; Thaler, R.; Weinberger, T.; Pick, R.; Mass, E.; Ishikawa-Ankerhold, H.C.; Margraf, A.; Hutter, S.; et al. Yolk Sac Macrophage Progenitors Traffic to the Embryo during Defined Stages of Development. Nat. Commun. 2018, 9, 75.
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Tremblay, M.-È.; Petratos, S.; Zoupi, L.; Boziki, M.; Kesidou, E.; Simeonidou, C.; Theotokis, P. Origin and Emergence of Microglia in the CNS—An Interesting (Hi)Story of an Eccentric Cell. Curr. Issues Mol. Biol. 2023, 45, 2609–2628.
- Shao, F.; Wang, X.; Wu, H.; Wu, Q.; Zhang, J. Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration. Front. Aging Neurosci. 2022, 14, 825086.
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare Coding Variants in PLCG2, ABI3, and TREM2 Implicate Microglial-Mediated Innate Immunity in Alzheimer’s Disease. Nat. Genet. 2017, 49, 1373–1384.
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.; Lucassen, P.J.; van Dam, A.-M. Microglial Phenotypes and Toll-like Receptor 2 in the Substantia Nigra and Hippocampus of Incidental Lewy Body Disease Cases and Parkinson’s Disease Patients. Acta Neuropathol. Commun. 2014, 2, 90.
- Smajić, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-Cell Sequencing of Human Midbrain Reveals Glial Activation and a Parkinson-Specific Neuronal State. Brain 2022, 145, 964–978.
- Prineas, J.W.; Kwon, E.E.; Cho, E.-S.; Sharer, L.R.; Barnett, M.H.; Oleszak, E.L.; Hoffman, B.; Morgan, B.P. Immunopathology of Secondary-Progressive Multiple Sclerosis. Ann. Neurol. 2001, 50, 646–657.
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘Homeostatic’ Microglia and Patterns of Their Activation in Active Multiple Sclerosis. Brain 2017, 140, 1900–1913.
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277.
- Bartholomäus, I.; Kawakami, N.; Odoardi, F.; Schläger, C.; Miljkovic, D.; Ellwart, J.W.; Klinkert, W.E.F.; Flügel-Koch, C.; Issekutz, T.B.; Wekerle, H.; et al. Effector T Cell Interactions with Meningeal Vascular Structures in Nascent Autoimmune CNS Lesions. Nature 2009, 462, 94–98.
- Wasser, B.; Luchtman, D.; Löffel, J.; Robohm, K.; Birkner, K.; Stroh, A.; Vogelaar, C.F.; Zipp, F.; Bittner, S. CNS-Localized Myeloid Cells Capture Living Invading T Cells during Neuroinflammation. J. Exp. Med. 2020, 217, e20190812.
- De Schepper, S.; Ge, J.Z.; Crowley, G.; Ferreira, L.S.S.; Garceau, D.; Toomey, C.E.; Sokolova, D.; Rueda-Carrasco, J.; Shin, S.-H.; Kim, J.-S.; et al. Perivascular Cells Induce Microglial Phagocytic States and Synaptic Engulfment via SPP1 in Mouse Models of Alzheimer’s Disease. Nat. Neurosci. 2023, 26, 406–415.
- Prinz, M.; Schmidt, H.; Mildner, A.; Knobeloch, K.-P.; Hanisch, U.-K.; Raasch, J.; Merkler, D.; Detje, C.; Gutcher, I.; Mages, J.; et al. Distinct and Nonredundant In Vivo Functions of IFNAR on Myeloid Cells Limit Autoimmunity in the Central Nervous System. Immunity 2008, 28, 675–686.
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Miró-Mur, F.; Pedragosa, J.; Zawadzka, M.; Pilanc, P.; Planas, A.M.; Kaminska, B. Defining Molecular Identity and Fates of CNS-Border Associated Macrophages after Ischemic Stroke in Rodents and Humans. Neurobiol. Dis. 2020, 137, 104722.
- Bechmann, I.; Priller, J.; Kovac, A.; Böntert, M.; Wehner, T.; Klett, F.F.; Bohsung, J.; Stuschke, M.; Dirnagl, U.; Nitsch, R. Immune Surveillance of Mouse Brain Perivascular Spaces by Blood-Borne Macrophages. Eur. J. Neurosci. 2001, 14, 1651–1658.
- Hickey, W.F.; Vass, K.; Lassmann, H. Bone Marrow-Derived Elements in the Central Nervous System: An Immunohistochemical and Ultrastructural Survey of Rat Chimeras. J. Neuropathol. Exp. Neurol. 1992, 51, 246–256.
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of Tissue-Resident Macrophages during Organogenesis. Science 2016, 353, aaf4238.
- Masuda, T.; Amann, L.; Monaco, G.; Sankowski, R.; Staszewski, O.; Krueger, M.; Del Gaudio, F.; He, L.; Paterson, N.; Nent, E.; et al. Specification of CNS Macrophage Subsets Occurs Postnatally in Defined Niches. Nature 2022, 604, 740–748.
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and Vertebral Bone Marrow Are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma. Science 2021, 373, eabf7844.
- Wu, X.; Saito, T.; Saido, T.C.; Barron, A.M.; Ruedl, C. Microglia and CD206+ Border-Associated Mouse Macrophages Maintain Their Embryonic Origin during Alzheimer’s Disease. eLife 2021, 10, e71879.
- Huang, G.; Zhang, P.; Hirai, H.; Elf, S.; Yan, X.; Chen, Z.; Koschmieder, S.; Okuno, Y.; Dayaram, T.; Growney, J.D.; et al. PU.1 Is a Major Downstream Target of AML1 (RUNX1) in Adult Mouse Hematopoiesis. Nat. Genet. 2008, 40, 51–60.
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia Emerge from Erythromyeloid Precursors via Pu.1- and Irf8-Dependent Pathways. Nat. Neurosci. 2013, 16, 273–280.
- Herbomel, P.; Thisse, B.; Thisse, C. Zebrafish Early Macrophages Colonize Cephalic Mesenchyme and Developing Brain, Retina, and Epidermis through a M-CSF Receptor-Dependent Invasive Process. Dev. Biol. 2001, 238, 274–288.
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a Unique TGF-β–Dependent Molecular and Functional Signature in Microglia. Nat. Neurosci. 2014, 17, 131–143.
- Ivan, D.C.; Berve, K.C.; Walthert, S.; Monaco, G.; Borst, K.; Bouillet, E.; Ferreira, F.; Lee, H.; Steudler, J.; Buch, T.; et al. Insulin-like Growth Factor-1 Receptor Controls the Function of CNS-Resident Macrophages and Their Contribution to Neuroinflammation. Acta Neuropathol. Commun. 2023, 11, 35.
- Hamilton, J.A.; Achuthan, A. Colony Stimulating Factors and Myeloid Cell Biology in Health and Disease. Trends Immunol. 2013, 34, 81–89.
- Hamilton, J.A. Colony-Stimulating Factors in Inflammation and Autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544.
- Sauter, K.A.; Bouhlel, M.A.; O’Neal, J.; Sester, D.P.; Tagoh, H.; Ingram, R.M.; Pridans, C.; Bonifer, C.; Hume, D.A. The Function of the Conserved Regulatory Element within the Second Intron of the Mammalian Csf1r Locus. PLoS ONE 2013, 8, e54935.
- Munro, D.A.D.; Bradford, B.M.; Mariani, S.A.; Hampton, D.W.; Vink, C.S.; Chandran, S.; Hume, D.A.; Pridans, C.; Priller, J. CNS Macrophages Differentially Rely on an Intronic Csf1r Enhancer for Their Development. Dev. Camb. Engl. 2020, 147, dev194449.
- Rojo, R.; Raper, A.; Ozdemir, D.D.; Lefevre, L.; Grabert, K.; Wollscheid-Lengeling, E.; Bradford, B.; Caruso, M.; Gazova, I.; Sánchez, A.; et al. Deletion of a Csf1r Enhancer Selectively Impacts CSF1R Expression and Development of Tissue Macrophage Populations. Nat. Commun. 2019, 10, 3215.
- Hagemeyer, N.; Kierdorf, K.; Frenzel, K.; Xue, J.; Ringelhan, M.; Abdullah, Z.; Godin, I.; Wieghofer, P.; Costa Jordão, M.J.; Ulas, T.; et al. Transcriptome-Based Profiling of Yolk Sac-Derived Macrophages Reveals a Role for Irf8 in Macrophage Maturation. EMBO J. 2016, 35, 1730–1744.
- Moura Silva, H.; Kitoko, J.Z.; Queiroz, C.P.; Kroehling, L.; Matheis, F.; Yang, K.L.; Reis, B.S.; Ren-Fielding, C.; Littman, D.R.; Bozza, M.T.; et al. C-MAF-Dependent Perivascular Macrophages Regulate Diet-Induced Metabolic Syndrome. Sci. Immunol. 2021, 6, eabg7506.
- Wang, Q.; Zhao, N.; Kennard, S.; Lilly, B. Notch2 and Notch3 Function Together to Regulate Vascular Smooth Muscle Development. PloS ONE 2012, 7, e37365.
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Miliaras, D.; Kesidou, E.; Boziki, M.; Petratos, S.; Grigoriadis, N.; Theotokis, P. Developmental Cues and Molecular Drivers in Myelinogenesis: Revisiting Early Life to Re-Evaluate the Integrity of CNS Myelin. Curr. Issues Mol. Biol. 2022, 44, 3208–3237.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069.
- Butovsky, O.; Weiner, H.L. Microglial Signatures and Their Role in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 622–635.
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698.
- Chen, Z.; Trapp, B.D. Microglia and Neuroprotection. J. Neurochem. 2016, 136 (Suppl. 1), 10–17.
- Goddery, E.N.; Fain, C.E.; Lipovsky, C.G.; Ayasoufi, K.; Yokanovich, L.T.; Malo, C.S.; Khadka, R.H.; Tritz, Z.P.; Jin, F.; Hansen, M.J.; et al. Microglia and Perivascular Macrophages Act as Antigen Presenting Cells to Promote CD8 T Cell Infiltration of the Brain. Front. Immunol. 2021, 12, 726421.
- Fabriek, B.O.; Van Haastert, E.S.; Galea, I.; Polfliet, M.M.J.; Döpp, E.D.; Van Den Heuvel, M.M.; Van Den Berg, T.K.; De Groot, C.J.A.; Van Der Valk, P.; Dijkstra, C.D. CD163-Positive Perivascular Macrophages in the Human CNS Express Molecules for Antigen Recognition and Presentation. Glia 2005, 51, 297–305.
- Mato, M.; Ookawara, S.; Sakamoto, A.; Aikawa, E.; Ogawa, T.; Mitsuhashi, U.; Masuzawa, T.; Suzuki, H.; Honda, M.; Yazaki, Y.; et al. Involvement of Specific Macrophage-Lineage Cells Surrounding Arterioles in Barrier and Scavenger Function in Brain Cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 3269–3274.
- Lim, H.Y.; Lim, S.Y.; Tan, C.K.; Thiam, C.H.; Goh, C.C.; Carbajo, D.; Chew, S.H.S.; See, P.; Chakarov, S.; Wang, X.N.; et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 2018, 49, 326–341.e7.
- He, H.; Mack, J.J.; Güç, E.; Warren, C.M.; Squadrito, M.L.; Kilarski, W.W.; Baer, C.; Freshman, R.D.; McDonald, A.I.; Ziyad, S.; et al. Perivascular Macrophages Limit Permeability. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2203–2212.
- Galanternik, M.V.; Castranova, D.; Gore, A.V.; Blewett, N.H.; Jung, H.M.; Stratman, A.N.; Kirby, M.R.; Iben, J.; Miller, M.F.; Kawakami, K.; et al. A Novel Perivascular Cell Population in the Zebrafish Brain. eLife 2017, 6, e24369.
- Liu, C.; Wu, C.; Yang, Q.; Gao, J.; Li, L.; Yang, D.; Luo, L. Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 2016, 44, 1162–1176.
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell 2016, 165, 882–895.
- Mato, M.; Ookawara, S.; Sano, M.; Fukuda, S. Uptake of Fat by Fluorescent Granular Perithelial Cells in Cerebral Cortex after Administration of Fat Rich Chow. Experientia 1982, 38, 1496–1498.
- Cummings, J. Alzheimer’s Disease Diagnostic Criteria: Practical Applications. Alzheimers Res. Ther. 2012, 4, 35.
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554.
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of Tau Protein in Alzheimer’s Disease: The Prime Pathological Player. Int. J. Biol. Macromol. 2020, 163, 1599–1617.
- Weber, S.A.; Patel, R.K.; Lutsep, H.L. Cerebral Amyloid Angiopathy: Diagnosis and Potential Therapies. Expert Rev. Neurother. 2018, 18, 503–513.
- Hawkes, C.A.; McLaurin, J. Selective Targeting of Perivascular Macrophages for Clearance of Beta-Amyloid in Cerebral Amyloid Angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266.
- Park, L.; Uekawa, K.; Garcia-Bonilla, L.; Koizumi, K.; Murphy, M.; Pistik, R.; Younkin, L.; Younkin, S.; Zhou, P.; Carlson, G.; et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ. Res. 2017, 121, 258–269.
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12.
- Lee, J.-W.; Chun, W.; Lee, H.J.; Kim, S.-M.; Min, J.-H.; Kim, D.-Y.; Kim, M.-O.; Ryu, H.W.; Lee, S.U. The Role of Microglia in the Development of Neurodegenerative Diseases. Biomedicines 2021, 9, 1449.
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease with the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356.
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial Exosomes Facilitate α-Synuclein Transmission in Parkinson’s Disease. Brain J. Neurol. 2020, 143, 1476–1497.
- Schonhoff, A.; Figge, D.; Williams, G.; Jurkuvenaite, A.; Gallups, N.; Childers, G.; Webster, J.; Standaert, D.; Goldman, J.; Harms, A. Border-Associated Macrophages Mediate the Neuroinflammatory Response in an Alpha-Synuclein Model of Parkinson Disease. bioRxiv 2022.
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H.; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against α-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. 2016, 36, 5144–5159.
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights from the Atlas of MS, Third Edition. Mult. Scler. J. 2020, 26, 1816–1821.
- Mirmosayyeb, O.; Brand, S.; Barzegar, M.; Afshari-Safavi, A.; Nehzat, N.; Shaygannejad, V.; Sadeghi Bahmani, D. Clinical Characteristics and Disability Progression of Early- and Late-Onset Multiple Sclerosis Compared to Adult-Onset Multiple Sclerosis. J. Clin. Med. 2020, 9, 1326.
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic Criteria for Multiple Sclerosis: 2010 Revisions to the McDonald Criteria. Ann. Neurol. 2011, 69, 292–302.
- Alcina, A.; Fedetz, M.; Vidal-Cobo, I.; Andrés-León, E.; García-Sánchez, M.-I.; Barroso-del-Jesus, A.; Eichau, S.; Gil-Varea, E.; Villar, L.-M.; Saiz, A.; et al. Identification of the Genetic Mechanism That Associates L3MBTL3 to Multiple Sclerosis. Hum. Mol. Genet. 2022, 31, 2155–2163.
- Jandric, D.; Lipp, I.; Paling, D.; Rog, D.; Castellazzi, G.; Haroon, H.; Parkes, L.; Parker, G.J.M.; Tomassini, V.; Muhlert, N. Mechanisms of Network Changes in Cognitive Impairment in Multiple Sclerosis. Neurology 2021, 97, e1886–e1897.
- Correale, J.; Marrodan, M.; Ysrraelit, M.C. Mechanisms of Neurodegeneration and Axonal Dysfunction in Progressive Multiple Sclerosis. Biomedicines 2019, 7, 14.
- Jäckle, K.; Zeis, T.; Schaeren-Wiemers, N.; Junker, A.; van der Meer, F.; Kramann, N.; Stadelmann, C.; Brück, W. Molecular Signature of Slowly Expanding Lesions in Progressive Multiple Sclerosis. Brain 2020, 143, 2073–2088.
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central Nervous System Macrophages in Progressive Multiple Sclerosis: Relationship to Neurodegeneration and Therapeutics. J. Neuroinflamm. 2022, 19, 45.
- Miedema, A.; Gerrits, E.; Brouwer, N.; Jiang, Q.; Kracht, L.; Meijer, M.; Nutma, E.; Peferoen-Baert, R.; Pijnacker, A.T.E.; Wesseling, E.M.; et al. Brain Macrophages Acquire Distinct Transcriptomes in Multiple Sclerosis Lesions and Normal Appearing White Matter. Acta Neuropathol. Commun. 2022, 10, 8.
- Locatelli, G.; Theodorou, D.; Kendirli, A.; Jordão, M.J.C.; Staszewski, O.; Phulphagar, K.; Cantuti-Castelvetri, L.; Dagkalis, A.; Bessis, A.; Simons, M.; et al. Mononuclear Phagocytes Locally Specify and Adapt Their Phenotype in a Multiple Sclerosis Model. Nat. Neurosci. 2018, 21, 1196–1208.
- Theotokis, P.; Lourbopoulos, A.; Touloumi, O.; Lagoudaki, R.; Kofidou, E.; Nousiopoulou, E.; Poulatsidou, K.-N.; Kesidou, E.; Tascos, N.; Spandou, E.; et al. Time Course and Spatial Profile of Nogo-A Expression in Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice. J. Neuropathol. Exp. Neurol. 2012, 71, 907–920.
- Theotokis, P.; Touloumi, O.; Lagoudaki, R.; Nousiopoulou, E.; Kesidou, E.; Siafis, S.; Tselios, T.; Lourbopoulos, A.; Karacostas, D.; Grigoriadis, N.; et al. Nogo Receptor Complex Expression Dynamics in the Inflammatory Foci of Central Nervous System Experimental Autoimmune Demyelination. J. Neuroinflamm. 2016, 13, 265.
- Montilla, A.; Zabala, A.; Er-Lukowiak, M.; Rissiek, B.; Magnus, T.; Rodriguez-Iglesias, N.; Sierra, A.; Matute, C.; Domercq, M. Microglia and Meningeal Macrophages Depletion Delays the Onset of Experimental Autoimmune Encephalomyelitis. Cell Death Dis. 2023, 14, 16.
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse Models of Multiple Sclerosis: Experimental Autoimmune Encephalomyelitis and Theiler’s Virus-Induced Demyelinating Disease. Methods Mol. Biol. 2012, 900, 381–401.
- Donninelli, G.; Saraf-Sinik, I.; Mazziotti, V.; Capone, A.; Grasso, M.G.; Battistini, L.; Reynolds, R.; Magliozzi, R.; Volpe, E. Interleukin-9 Regulates Macrophage Activation in the Progressive Multiple Sclerosis Brain. J. Neuroinflamm. 2020, 17, 149.
- Roczkowsky, A.; Doan, M.A.L.; Hlavay, B.; Mamik, M.K.; Branton, W.G.; McKenzie, B.A.; Saito, L.B.; Schmitt, L.; Eitzen, G.; Di Cara, F.; et al. Peroxisome Injury in Multiple Sclerosis: Protective Effects of 4-Phenylbutyrate in CNS-Associated Macrophages. J. Neurosci. Off. J. Soc. Neurosci. 2022, 42, 7152–7165.
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The Physiology of Foamy Phagocytes in Multiple Sclerosis. Acta Neuropathol. Commun. 2018, 6, 124.
- Haidar, M.; Loix, M.; Vanherle, S.; Dierckx, T.; Vangansewinkel, T.; Gervois, P.; Wolfs, E.; Lambrichts, I.; Bogie, J.F.J.; Hendriks, J.J.A. Targeting Lipophagy in Macrophages Improves Repair in Multiple Sclerosis. Autophagy 2022, 18, 2697–2710.
- Pedragosa, J.; Salas-Perdomo, A.; Gallizioli, M.; Cugota, R.; Miró-Mur, F.; Briansó, F.; Justicia, C.; Pérez-Asensio, F.; Marquez-Kisinousky, L.; Urra, X.; et al. CNS-Border Associated Macrophages Respond to Acute Ischemic Stroke Attracting Granulocytes and Promoting Vascular Leakage. Acta Neuropathol. Commun. 2018, 6, 76.
- Wan, H.; Brathwaite, S.; Ai, J.; Hynynen, K.; Macdonald, R.L. Role of Perivascular and Meningeal Macrophages in Outcome following Experimental Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2021, 41, 1842–1857.
- Kolosowska, N.; Keuters, M.H.; Wojciechowski, S.; Keksa-Goldsteine, V.; Laine, M.; Malm, T.; Goldsteins, G.; Koistinaho, J.; Dhungana, H. Peripheral Administration of IL-13 Induces Anti-Inflammatory Microglial/Macrophage Responses and Provides Neuroprotection in Ischemic Stroke. Neurotherapeutics 2019, 16, 1304–1319.