 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Frank Friedrich Gellrich | + 4616 word(s) | 4616 | 2021-04-27 10:21:49 | | | |
| 2 | Peter Tang | Meta information modification | 4616 | 2021-05-17 09:52:16 | | |
Video Upload Options
Malignant melanoma is more dangerous than most other skin cancers due to its ability to spread early and aggressively. Until the development of new therapeutic strategies, the median survival of patients with metastatic melanoma was just a few months. Immunotherapy, the first regimen, leading to significant improvement, blocks immune checkpoints, which normally dampen immune responses, enabling our defense cells to recognize and destroy cancer cells again. Immunotherapy achieves long-term survival in about 50% of metastatic melanoma patients. Besides, targeted therapy has also significantly improved the survival of melanoma patients, blocking cell-signaling proteins, which are altered in about 50% of melanomas and lead to uncontrolled tumor cell growth. In addition to the approved regimens, there are numerous new treatment strategies, ranging from modified viruses to personalized immune cells that attack and destroy tumor cells.
1. Introduction
Around 5–6 decades ago, a new ideal of beauty in skin color became more and more apparent: tanned skin reflecting a sporty and healthy lifestyle. Thereby increased UV-exposition is regarded as the most important reason that incidence rates of melanoma keep rising dramatically over the past years. Although being far less prevalent than other skin malignancies in total, melanoma is responsible for around 90% of deaths concerning skin cancer.
Melanoma arises from atypically transformed melanocytes whose fetal precursor cells originate from the neural crest. Like other cell types, melanocytes undergo a stepwise process acquiring features summarized as the hallmarks of cancer [1][2]: 1. Self-sufficiency in growth signals; 2. Insensitivity to growth-inhibitory signals; 3. Evasion of programmed cell death; 4. Limitless replicative potential; 5. Sustained angiogenesis; 6. Tissue invasion and metastasis; 7. Deregulation of cellular energetics; 8. Avoiding immune destruction. These pathogenic changes frequently develop from activation of tumor-promoting oncogenes and/or silencing of tumor-suppressor genes by somatic mutations, genomic rearrangements (deletion, amplification), or dysregulated gene expression through epigenetic mechanisms. Mutations in B-Raf protooncogene, serine/threonine kinase (BRAF), NRAS protooncogene GTPase (NRAS), or KIT protooncogene, receptor tyrosine kinase (KIT) genes, as well as loss of tumor suppressor phosphatase and tensin homolog (PTEN) or cyclin-dependent kinase inhibitor 2A (CDKN2A) occur at early stages of malignant transformation [3]. However, loss of E-Cadherin and upregulation of N-Cadherin are characteristic for later stages [3]. The changes associated with the cellular adhesion molecules E-Cadherin and N-Cadherin are part of the epithelial to mesenchymal transition (EMT) and substantially contribute to progression of melanoma cells by promoting detachment from the basal keratinocytes within the epidermis (invasion) and subsequent intravasation into the blood stream (metastasis). Genetic instability and external factors such as ultraviolet (UV)-radiation may accelerate malignant transformation and usually affect relevant survival pathways. In case of melanoma, the most frequently hyperactivated pathways are the mitogen-activated protein kinase (MAPK) and the phosphatidylinositol 3-kinase (PI3K) pathway [4][5][6][7]. However, there is evidence of other pathways being hijacked as well, including but not limited to hepatocyte growth factor (HGF)- or microphthalmia-associated transcription factor (MITF)-signaling [8][9][10][11], for example. In summary, specific sequential steps are required for melanocytes to transform into malignant melanoma and metastasize to lymph nodes and distant organs like the lung, the liver, or the brain. Consequently, each event may worsen patient prognosis, emphasizing the importance of early diagnosis for improved patient outcome.
Until recently, the diagnosis of distant metastases had an unfavorable prognosis with a median survival of 6–9 months [12][13]. The ground-breaking discovery of immunotherapy and targeted therapy to be suitable defense mechanisms revolutionized the standard of care and led to new optimism in treating melanoma. Besides the already existing regimens, there is a variety of promising approaches for new therapeutic options.
2. Immune Checkpoint Inhibitors
In 1995, James P. Allison and colleagues revealed the purpose of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), an immune checkpoint molecule, which is able to downregulate steps in T cell activation [14][15]. It has been hypothesized that, by blocking these inhibitory immune checkpoint molecules, it is possible to stimulate the immune response against cancer cells.
Ipilimumab, a monoclonal antibody against CTLA-4 (Figure 1), was the first drug to demonstrate a significant improvement in OS of advanced melanoma in a phase 3 study (Table 1) [16]. Over time, however, several trials demonstrated that ipilimumab monotherapy is inferior to antibodies directed against programmed death 1 (PD-1) receptor [17][18][19][20].
Besides blocking the inhibiting immune checkpoint CTLA-4 and thereby bolstering immune response against cancer cells, a second target became apparent in the last decades: In 1992, Tasuku Honjo and colleagues discovered the programmed death 1 (PD-1) receptor [21], expressed on a variety of immune cells, such as T cells, B cells, monocytes, dendritic cells, and tumor-infiltrating lymphocytes (TILs).
PD-1, interacting with its ligand PD-L1 on tumor cells and antigen-presenting cells, and PD-L2 on activated monocytes and dendritic cells, inhibits T cell activity, thereby regulating immune tolerance. Physiologically, the PD-1/PD-L1 pathway is needed as a regulatory checkpoint in order to slow down the degree of inflammation. Various tumors, especially melanomas, overexpress PD-L1 to escape from the inflammatory process and generate an immunosuppressive tumor environment. Blocking either PD-1 or PD-L1 with antibodies facilitates an effective antitumor immune response to kill tumor cells.
The discovery of immune checkpoint blockade led to a new era in fighting cancer. To appreciate this ground-breaking discovery, the Nobel Prize for Physiology or Medicine was awarded jointly to James P. Allison and Tasuku Honjo in 2018.
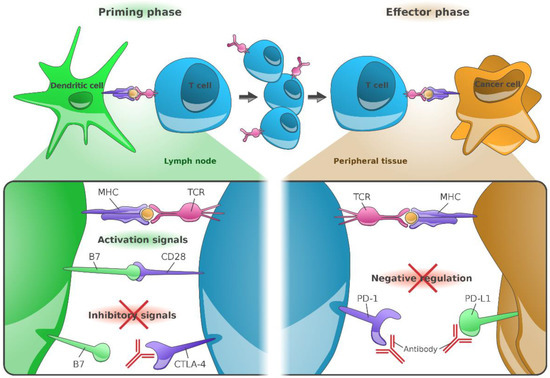
Figure 1. Immunological mode of action of anti-CTLA-4 (CD152), anti-PD-1 (CD279), and anti-PD-L1 (CD274) monoclonal antibodies. The major histocompatibility complex (MHC), present on the surface of cancer cells or dendritic cells, presents peptides derived from tumor-associated antigens (TAAs), which are recognized by T-cells via their T-cell receptors (TCR). Additional cell signaling is provided by the co-stimulatory molecules B7-1 (CD80) or B7-2 (CD86). Both factors are required for T-cell priming. Once activated, T-cells upregulate CTLA-4 expression on their cell surface; in contrast, binding of CTLA-4 to B7 receptors of dendritic cells results into inhibition of T-cell activation. Anti-CTLA-4 directed antibodies hence block inhibitory signaling and restore T-cell activation in lymph nodes. Continued stimulation results into upregulation of PD-1 receptors in T-cells, their (parallel) inhibition prevents the interaction of PD-1 with its ligand, PD-L1. Due to the omission of such negative regulation, tumor cells expressing PD-L1 can again be identified by effector T-cells [22]. Figure adapted from Ribas A [22] and created by Gellrich FF, first published in J. Clin. Med. [23].
Table 1. Relevant clinical trials demonstrating efficacy of immune checkpoint inhibitors and BRAF kinase inhibitors and MEK kinase inhibitors (BRAF/MEK inhibitors) in unresectable/metastatic melanoma.
|
Trial (NCT n°) |
Agents/dose (mg/kg or mg/m2) |
Phase |
Patients |
N |
Prim. EP |
ORR, % |
PFS Med, mts (HR [95%CI]) |
OS Med, mts (HR [95%CI]) |
Ref. |
|---|---|---|---|---|---|---|---|---|---|
|
Anti-CTLA-4 directed Immunotherapy |
|||||||||
|
MDX010-020 (NCT00094653) |
Ipi 3 + gp100 vs. Ipi 3 vs. gp100 (3:1:1) |
III |
Unresectable Stage III/IV, pre-treated, HLA-A*0201–pos. |
676 |
OS |
6 vs. 11 vs. 2 |
2.8 vs. 2.9 vs. 2.8 (0.64 [0.50–0.83]) |
10.0 vs. 10.1 vs. 6.4 (0.66 [0.51–0.87] a |
[16] |
|
Anti-PD-1-directed Immunotherapy (Immune Checkpoint Inhibition mono, or in combination with an anti-CTLA-4 antagonist) |
|||||||||
|
CM-066 (NCT01721772) |
Niv 3 q2w + Placebo vs. Placebo + Dacarbazine 1000 q3w (1:1) |
III |
Metastatic, untreated, BRAF WT |
418 |
OS |
40 vs. 14 |
5.1 vs. 2.2 (0.43 [0.34–0.56]) |
37.5 vs. 11.2 (0.46 [0.36–0.59]) |
|
|
CM-067 (NCT01844505) |
Niv 1 +Ipi 3 (q3w) x4 → Niv 3; Niv 3 alone q2w, vs. Ipi 3 q3w x4 (1:1:1) |
III |
Unresectable Stage III/IV, untreated |
945 |
PFS and OS (co-primary) |
58 vs. 44 vs. 19 |
11.5 vs. 6.9 vs. vs. 2.9 (0.42 b [0.31–0.57]) c; (0.57 b [0.43–0.76]) d |
||
|
CM-511 (NCT02714218) |
Niv 1 +Ipi 3 (q3w) x4 → Niv 3 vs. Niv 3 +Ipi 1 (q3w) x4 → Niv 3 (1:1) |
III |
Unresectable Stage III/IV, untreated |
360 |
TRAE rate (grade 3–5) |
TRAE: 48 vs. 34 |
8.9 vs. 9.9 (1.06 [0.79–1.42]) |
NR vs. NR (1.09 [0.73–1.62]) |
[28] |
|
CM-003 (NCT00730639) |
Niv 0.1 vs. Niv 0.3 vs. Niv 1 vs. Niv 3 vs. Niv 10 (all q2w) |
I |
1–5 prior systemic therapies (ocular melanoma allowed) |
107 |
Safety, ORR |
35 vs. 29 vs. 31 vs. 41 vs. 20 |
3.6 vs. 1.9 vs. 9.1 vs. 9.7 vs. 3.7 |
16.2 vs. 12.5 vs. 25.3 vs. 20.3 vs. 11.7 |
[29] |
|
KN-006 (NCT1866319) |
Pem 10 q2w vs. Pem 10 q3w vs. Ipi 3 q3w (1:1:1) |
III |
Unresectable Stage III/IV, ≤1 prior systemic therapy, Ipi-naïve |
834 |
PFS & OS (co-primary) |
34 vs. 33 vs. 12 |
8.4 vs. 3.4 (0.57 [0.48–0.67]) e |
32.7 vs. 15.9 (0.73 [0.61–0.88]) e |
[30] |
|
KN-001 (NCT01295827) |
Pem 2 q3w/Pem 10 q3w/Pem 10 q2w |
IB |
Advanced or metastatic, pre- or untreated |
655 |
ORR |
41 (52, if untreated) |
8.3 (16.9 in the 151 untreated patients) |
23.8 (38.6 in the 151 untreated patients) |
[31] |
|
Herpes simplex virus-1 derived Oncolytic virus |
|||||||||
|
OPTiM (NCT00769704) |
Tal (106 pfu/mL →106 pfu/mL q3w i.l.) vs. GM-CSF 125 µg/m2 (2:1) |
Unresectable Stage IIIB/IV M1c, ≥1 sub-/cutaneous lesion 10 mm |
436 |
DRR (16 vs. 2) |
DRR: 19% vs. 1% |
nr |
23.3 vs. 18.9 (0.79 [0.62–1.00]) |
[32] |
|
|
Targeted therapy (BRAF mono) 16 vs. 2 |
|||||||||
|
BRIM-3 (NCT01006980) |
Vem 960 mg td vs. DTIC (1:1) |
III |
Metastatic, untreated, BRAFV600E-mut. |
675 |
PFS & OS (co-primary) |
48 vs. 5 |
5.3 vs. 1.6 (0.26 [0.20–0.33]) |
NR vs. 7.9 (0.37 [0.26–0.55]) |
[33] |
|
Combined BRAF+MEK blockade |
|||||||||
|
COMBI-v (NCT01597908) |
Dab 150 bd + Tra 2 od vs. Vem 960 bd (1:1) |
III |
Metastatic, untreated, BRAFV600E/K-mut. |
704 |
OS |
64 vs. 51 |
11.4 vs. 7.3 (0.56 [0.46–0.69]) |
NR vs. 17.2 (0.69 [0.53–0.69]) |
|
|
COMBI-d (NCT01584648) |
Dab 150 bd + Tra 2 od vs. Dab 150 bd (1:1) |
III |
Unresectable Stage IIIC/IV, untreated, BRAFV600E/K-mut. |
423 |
PFS |
69 vs. 53 |
11.0 vs. 8.8 (0.67 [0.53–0.84]) |
25.1 vs. 18.7 (0.71 [0.55–0.92]) |
|
|
CoBRIM (NCT01689519) |
Cob 60 od d1-21 + Vem 960 bd vs. Vem 960 bd + Placebo (1:1) |
III |
Unresectable Stage IIIB–IV, untreated, BRAF V600-mut. |
495 |
PFS |
68 vs. 45 |
9.9 vs. 6.2 (0.51 [0.39–0.68]) |
22.3 vs. 17.4 (0.70 [0.55–0.90]) |
|
|
COLUMBUS (NCT01909453) |
Enc 450 od + Bin 45 mg bd vs. Enc 300 mg od vs. Vem 960 mg bd (1:1:1) |
III |
Unresectable Stage IIIB–IV, un-treated (or progressed after first-line IT), BRAFV600E/K-mut. |
577 |
PFS |
63 vs.51 vs. 40 |
14.9 vs. × 9.6 vs. 7.3 (0.54 [0.41–0.71] f |
33.6 vs. 23.5 vs. 16.9 (0.61 [0.47–0.79] f |
|
|
NEMO (NCT01763164) |
Bin 45 bd vs. Dacarbazine 1000 |
III |
Unresectable Stage IIIB–IV, un-treated (or progressed after first-line IT), NRASV600E/K-mut. |
402 |
PFS |
15 vs. 7 |
2.8 vs. 1.5 (0.62 [0.47–0.80]) |
11.0 vs. 10.1 (1.00 [0.75–1.33]) |
[41] |
|
Triplett therapy (ICI + BRAF/MEK-blockade) |
|||||||||
|
IMSpire150 (NCT02908672) |
Ate 840 d1,15 + Vem 720 bd + Cob 60 od d1-21 vs. Placebo + Vem 960 bd + Cob 60 od d1-21 (all: q4w) |
III |
Untreated, BRAF-mut. |
514 |
PFS |
66 vs. 65 |
15.1 vs. 10.6 (0.78 [0.63–0.97]) |
not yet reached/reported |
[42] |
|
COMBI-I (NCT02967692) |
Spa 400 mg + Dab 150 bd + Tra 2 od vs. Placebo + Dab 150 + Tra 2 (q4w) |
III |
Unresectable Stage IIIB–IV, untreated, BRAF V600-mut. |
532 |
PFS |
16.2 vs. 12.0 (0.82 [0.66–1.03]) |
NR vs. NR (0.79 [0.59–1.05]) |
[43] |
|
a i.e., for the comparison of Ipi vs. gp100. b 99.5%CI. c i.e., for comparison of Niv + Ipi vs. Ipi. d i.e., for comparison of Niv + Ipi vs. Ipi. e i.e., for comparison of Pem (q2w + q3w arms) vs. Ipi. f i.e., for comparison of Enc + Bin vs. Vem. Abbreviations: →, then (drug) administered until disease progression or unacceptable toxicity; Ate, atezolizumab; bd, twice daily; Bin, binimetinib; Cob, cobimetinib; Dab, dabrafenib; Enc, encorafenib; EP, endpoint; i.l., intralesional; Ipi, ipilimumab; IT, immunotherapy; Niv, nivolumab; NR, not reached; nr, not reported; od, once daily; ORR, overall response rate; OS, median overall survival; Pem, pembrolizumab; PFS, median progression-free survival; q2w/q3w/q4w, all two/three/four weeks; Spa; spartalizumab; Tal, talimogene laherparepvec; Tra, trametinib; TRAE, treatment-related adverse events; Vem, vemurafenib; vs., versus.
3. T-VEC
Talimogene laherparepvec (T-VEC), a genetically modified herpes simplex virus of type 1 (Figure 2), is the first oncolytic virus therapy approved by the U.S. FDA and the EMA for use as a local treatment of unresectable melanoma of stage IIIB, IIIC, or IVM1a.
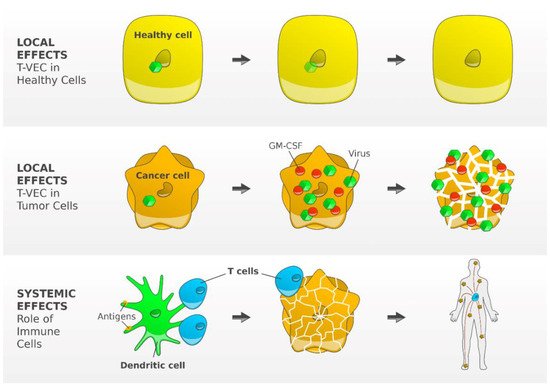
Figure 2. Mode of action of T-VEC, a genetically modified type 1 herpes simplex virus. The functional deletion of one of the two excised, non-essential viral genes, the gene herpesvirus neurovirulence factor (ICP34.5) is attenuating viral pathogenicity and thereby enhancing tumor-selective replication. Inside a normal cell, the virus cannot replicate but may replicate in tumor cells, inducing granulocyte-macrophage colony-stimulating factor (GM-CSF). Subsequent tumor cell lysis results into release of the virus, GM-CSF, and TAAs, which trigger the activity of dendritic cells resulting finally in an efficient induction and activation of tumor-reactive T cells [44]. Figure adapted from Andtbacka R [44] and created by Gellrich FF, first published in J. Clin. Med. [23].
Due to deletion of the infected cell protein (ICP) 34.5 gene, which enables neurovirulence, the virus has decreased pathogenicity with promoted selective replication in tumor cells [45]. Immunogenicity is supported by several factors. Deletion of the viral ICP47 gene gives antigenic viral and tumor-associated peptides access to MHC class I complexes, leading to reinforced host immune responses. Insertion of two copies of the human GM-CSF gene attracts local dendritic cells [46]. After injection, T-VEC is expected to replicate within the infected tumor cell. The subsequent lysis releases various fragments, e.g., tumor-associated antigens and GM-CSF, which initiate a local immune response and enhance dendritic cell migration. The dendritic cells trigger a systemic T cell response by presenting antigens to specific CD4+ cells and CD8+ cells in the regional lymph node [47].
The OPTiM trial enrolled 436 patients with injectable but not surgically resectable melanoma metastases either receiving intralesional T-VEC or subcutaneous GM-CSF (Table 1) [32][44][48][49]. The durable response rate, primary endpoint of the trial, was significantly higher in patients treated with T-VEC (19% vs. 1%). The most common TRAEs observed with T-VEC were influenza-like illness, pyrexia, chills, fatigue, and nausea. Grade 3 or 4 TRAEs were reported in 11% of patients treated with T-VEC and in 5% of patients treated with GM-CSF.
Data of 21 patients in phase 1b of the MASTERKEY-265/KEYNOTE-034 study, investigating the combination of T-VEC with pembrolizumab in patients with metastatic melanoma, were presented at ASCO 2016 [50]. After a median follow-up of 33 weeks, the confirmed ORR was 48%, and the unconfirmed ORR was 57%. Grade 3 or 4 TRAEs occurred in 33% of patients. Recruitment of the phase 3 of the MASTERKEY-265/KEYNOTE-034 study has been terminated and the primary completion date is estimated in 2022.
4. Targeted Therapy
The discovery of mutations of the BRAF gene in human cancer cells in 2002 laid the groundwork for the targeted therapy of melanoma [51]. Activating mutations in BRAF are found in 40–50% of melanomas (Figure 3). In 2011, a randomized phase 3 trial showed the superiority of the BRAF inhibitor vemurafenib over dacarbazine in OS, PFS, and ORR, and was approved for the treatment of unresectable or metastatic BRAFV600E-mutated melanoma (Table 1) [33]. Although monotherapy with the BRAF inhibitor vemurafenib improves OS, responses are often short-lived. Following the discovery of the clinical activity of single-agent MEK inhibition [52], the use of BRAF/MEK inhibitor combinations was evaluated in clinical studies.
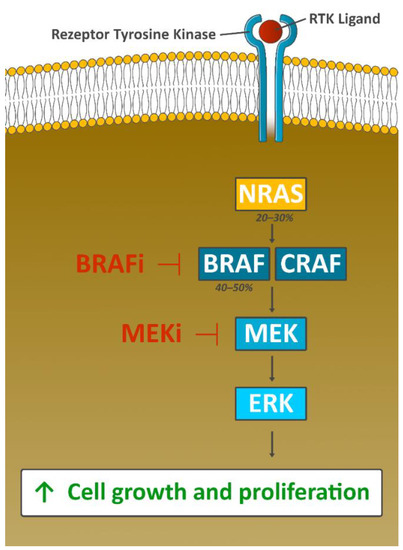
Figure 3. Oncogenic function of the MAPK signaling pathway. Genetic aberrations in this pathway are found in a vast majority of melanomas, with driver mutations in BRAF (V600E or V600K) occurring most often. The activation of BRAF results into phosphorylation of MEK and the activation of downstream MAP kinases like ERK. The pathway co-regulates tumor cell proliferation and cell survival; inhibition of BRAF downregulates the oncogenic function of MAPK signaling [33]. However, resistance to BRAF therapy is observed in about 50% of BRAF-mutated patients within 6–7 months after start of therapy. The CRAF-mediated reactivation of MAPK signaling pathway can be effectively blocked by the additional use of a MEK inhibitor (combined BRAF-MEK blockade), prolonging time to resistance development considerably. Consequently, anti-BRAF directed monotherapy is no longer used today. Figure adapted from Jenkins RW [53] and created byGellrich FF.
5. Sequencing and Combinations of BRAF/MEK Inhibitors and Immune Checkpoint Inhibitors
5.1. Mode of Action
Combined BRAF/MEK blockade results in rapid and robust disease control rates and remarkable ORR in BRAFV6oo-mutated metastatic melanoma. However, the DOR is limited in most patients, due to multiple mechanisms of acquired resistance [54]. In contrast, ICIs act more slowly, and sometimes responses are seen after a period of transitional progression (‘pseudo-progression’ due to the massive infiltration of T-cells into the tumor). ICIs induce less frequent but durable responses in approximately 50% of patients [55]. Intriguingly, besides their effect on the MAPK cell signaling pathway, BRAF/MEK inhibitors are thought to also address targets in the cancer-immunity cycle [56][57]. They (1) trigger the release of tumor cell antigens, (2) create an immune stimulatory microenvironment by increasing expression of immune stimulatory molecules and by reducing immunosuppressive cells and cytokines, (3) decrease VEGF expression and enhance infiltration of T cells into the tumor, (4) promote recognition of tumor cells by the immune system through increasing melanoma antigen and HLA I and/or HLA II expression, and (5) intensify reactivity and cytotoxicity of T cells [56]. Results from experimental medicine support the approach to sequence or combine targeted therapy and immunotherapy to trigger synergistic tumor control resulting in rapid, deep, and lasting responses.
5.2. First-Line BRAF/MEK Inhibitors versus Immune Checkpoint Inhibitors
Little is known about the best treatment sequence of BRAF/MEK inhibitors and ICIs.
A recent exploratory analysis of survival data from clinical trials with BRAF/MEK inhibitors and ICIs in patients with metastatic melanoma [58] demonstrated a superiority of BRAF/MEK inhibitors within the first 12 months, later changing to a superiority of anti-PD-1 alone or in combination with anti-CTLA-4 with 3-year OS rates of 41% for BRAF/MEK inhibitors, 50% for anti-PD-1 alone, and 58% for anti-PD-1 plus anti-CTLA-4. This observation reflects the primary resistance under ICIs, the acquired resistance under BRAF/MEK inhibitors and the higher frequency of durable responses under ICI treatment.
According to the ESMO consensus conference [20], first-line decisions between BRAF/MEK inhibitors or ICIs should be individualized and based on performance status, organs involved, LDH level, tumor burden, tumor progression kinetics, comorbidities, patient preference, and treatment goals (short-term versus long-term benefit). For patients requiring urgent treatment, the rapid clinical response observed with BRAF/MEK inhibitors may provide rapid disease control. Furthermore, the immunological effects of BRAF/MEK inhibitors may offer a chance for an early switch to ICIs [59]. Patients who can be treated with ICIs for the first few months should be considered for ICIs first, providing the chance for long-term disease control even after treatment discontinuation. Overall, the optimal sequencing strategy is yet to be established.
5.3. Novel Sequencing Approaches for BRAF/MEK Inhibitors and Immune Checkpoint Inhibitors
To study sequencing approaches, prospectively planned clinical trials randomize patients in advance to pre-defined sequences of systemic therapies (e.g., SECOMBIT, NCT02631447 and EBIN, NCT03235245).
In the ongoing phase 3 DREAMseq trial, patients with unresectable stage III or IV BRAFV600-mutant melanoma receive either BRAF/MEK inhibitor therapy with dabrafenib and trametinib and switch after progression to ICI therapy with ipilimumab and nivolumab, or the other way around [60]. The completion of the trial is estimated in 2022.
The phase 2 ImmunoCobiVem trial enrolled patients with unresectable or metastatic BRAFV600 mutant melanoma to receive BRAF/MEK inhibitor therapy with vemurafenib and cobimetinib for three months [61]. All patients who did not show disease progression were randomized to continue with vemurafenib and cobimetinib until PD, followed by anti-PD-L1 therapy with atezolizumab (arm A), or to receive atezolizumab until PD, followed by vemurafenib and cobimetinib (arm B). Results of these trials are eagerly awaited.
5.4. Novel Combinations of BRAF/MEK Inhibitors and Immune Checkpoint Inhibitors
Given the rapid and deep responses seen with BRAF/MEK inhibitors and the durable responses observed with ICIs, the combination of these therapeutic strategies appears promising. The phase 3 IMspire 150 trial (NCT02908672) combined atezolizumab, an anti-PD-L1 monoclonal antibody, with the BRAF/MEK inhibitor combination vemurafenib and cobimetinib in patients with BRAFV600-mutant metastatic melanoma [42]. Further, 514 patients were randomly assigned to receive atezolizumab, vemurafenib and cobimetinib or placebo, vemurafenib and cobimetinib, as first-line therapy. The study met its primary endpoint of PFS. Although there was no difference in the PFS curves for the first 6–8 months, the curves separated thereafter. The triple therapy did not increase the ORR (Table 1). Grade 3 or 4 TRAEs occurred in 79% of patients treated with the triple combination and in 73% of patients treated with vemurafenib plus cobimetinib only. In conclusion, the addition of atezolizumab to targeted therapy with vemurafenib and cobimetinib was tolerable and significantly increased PFS in patients with BRAFV600-mutant metastatic melanoma.
A triple therapy of the novel anti-PD-1-directed monoclonal antibody spartalizumab in combination with dabrafenib and trametinib was studied in the phase 3 COMBI-I trial (NCT02967692), compared to dabrafenib and trametinib plus placebo, as first-line therapy in patients with BRAFV600E/K-mutant unresectable (stage IIIC) or metastatic (stage IV) melanoma (Table 1) [43]. With an ORR for the triplet therapy being slightly higher (69%) compared to the doublet (64%), the trial unexpectedly did not meet its primary endpoint of investigator-assessed PFS for patients treated with the triple therapy (Table 1), although median PFS for the triplet was 4.2 months longer than for the doublet. TRAEs were however considerably higher for the triplet: grade 3 or 4 TRAEs occurred in 55% of patients in the triple therapy arm compared to 33% in the dabrafenib/trametinib arm; TRAEs leading to discontinuation of all study drugs occurred in 12% compared to 8% of patients, respectively.
In order to reduce the increased risk of toxicity when continuously combining ICIs with BRAF/MEK inhibitors, the phase 2b trial IMPemBra investigated the clinical benefit of intermittent, short-term BRAF/MEK inhibition during anti-PD-1 therapy—based on preclinical findings that short-term, but not long-term, BRAF/MEK inhibition induced strong T cell infiltration, acting hence synergistically with anti-PD-1 monoclonal antibodies [62]. Thiry-two patients with BRAFV600-mutant metastatic melanoma received pembrolizumab and were randomized in week 6 either to continue pembrolizumab monotherapy (cohort 1) or to add intermittent dabrafenib and trametinib 2 × 1 week (cohort 2) or 2 × 2 weeks (cohort 3) or continuously for 6 weeks (cohort 4). ORRs at week 18 were 62%, 75%, 75%, and 50% in cohort 1, 2, 3, and 4, respectively. Grade 3 or 4 TRAEs were reported in 12%, 12%, 50%, and 62% of patients. The authors concluded that the combination of pembrolizumab plus intermittent BRAF/MEK blockade with dabrafenib and trametinib might be more feasible and tolerable than continuous triple therapy.
5.5. Combination of Targeted Therapy with ICIs in BRAF-Wildtype Melanoma
There is some pre-clinical evidence that in BRAF-wildtype melanoma, the combination of MEK inhibitors with ICIs may enhance anti-tumor effects. Based on these findings, IMspire 170, a phase 3 trial, randomized 446 patients with unresectable stage III/IV BRAF-wildtype melanoma to receive either the MEK inhibitor cobimetinib plus atezolizumab, or the anti-PD-1 monoclonal antibody pembrolizumab alone [63]. The trial did not meet its primary endpoint, showing a median PFS of 5.5 months with cobimetinib plus atezolizumab vs. 5.7 months with pembrolizumab alone. Regarding the safety profiles, grade 3 or 4 TRAEs and discontinuation of treatment occurred in 67% and 12% of patients receiving the combined therapy versus 33% and 6% receiving pembrolizumab monotherapy.
6. Novel Drugs and Combinations
6.1. Introduction
Besides the already existing regimens, there is a variety of novel promising treatment strategies for patients with metastatic melanoma. Most of them aim to elicit an immune response against cancer cells. For example, NKTR-214 is a prodrug of IL-2 that stimulates expansion of CD8+ T cells, CD4+ T cells, and natural killer cells. IMCgp100 is a so-called ImmTAC (immune-mobilizing monoclonal T cell receptor against cancer) and acts as a link to redirect T cells to melanoma cells. In adoptive T cell transfer, a large number of antitumor lymphocytes is produced in vitro. After lymphodepletion, they are meant to serve as a highly personalized cancer therapy.
Combination of anti-PD-1 monoclonal antibodies with pegylated engineered interleukin-2 (IL-2).
In 1984, a 33-year-old woman, suffering from metastatic melanoma and progressing despite multiple prior therapies, was the first one receiving an infusion of aldesleukin (rIL-2), a recombinant version of IL-2. Only a few months later, the patient was free of disease. Being in CR over the years, this woman was the first to prove that it is possible to cure cancer by bolstering the patient’s immune system [64]. Later, aldesleukin was approved by the FDA as a treatment for metastatic melanoma based on pooled results of eight single-arm phase 2 trials. Interleukin had resulted—despite major toxicity concerns— in long-term survival in some patients, with “tail-of-the-Kaplan–Meier-curve” efficacy of a few percent, i.e., demonstrating long term recurrence-free survival. IL-2 leads to potent activation of immune cells, and in particular, to expansion of CD8+ T cells. Nevertheless, due to frequent and unchecked induction of the IL-2 cascade, this procedure is associated with serious AEs, e.g., hypotension, dyspnea, renal insufficiency, hepatic dysfunction, cardiac arrhythmias, and neurotoxicity [65][66].
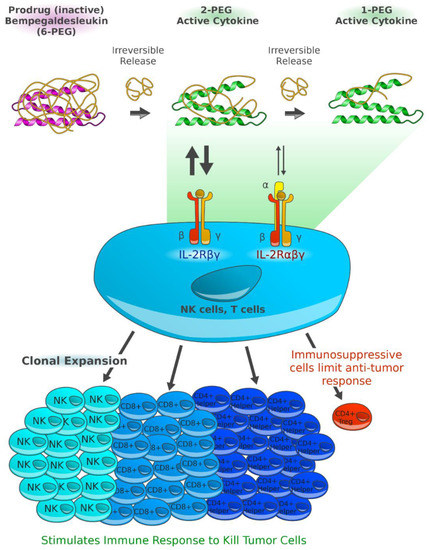
Today, next-generation aldesleukin-derivates like bempegaldesleukin (BEMPEG, NKTR-214), a human recombinant prodrug of IL-2, are under clinical investigation. BEMPEG is based on the IL-2 core with 6 releasable polyethylene glycol (PEG) chains attached. With fully pegylated IL-2 hardly showing biological activity, the partial pegylation and slow release of PEG chains, as demonstrated in vivo, leads to the formation of IL-2 conjugates, selectively stimulating CD8+ T cells, CD4+ T cells, and natural killer cells without increasing regulatory T cells within the tumor microenvironment [67]. Studies indicate that BEMPEG also leads to overexpression of ICOS, PD-1, and CTLA-4, implying an activation of immune cells [68]. Strong synergistic effects were demonstrated in mouse tumor models for the combination of NKTR-214 and an anti-CTLA-4 monoclonal antibody [67].
In the phase 1 PIVOT-02 trial, 38 immunotherapy-naïve patients with solid tumors including melanoma received BEMPEG and nivolumab [69]. The combination achieved an ORR of 59.5%, with 18.9% CRs. Flu-like symptoms (86.8%), rash (78.9%), fatigue (73.7%), and pruritus (52.6%) were common adverse events with the combination; for 21.1% of patients, grade 3 or 4 TRAEs were reported. Changes in immune signatures characteristic for treatment response, e.g., an accumulation of interferon-γ or CD8+ TILs, were demonstrated by biomarker analysis of baseline tumor biopsies. Responses were also observed in patients with a tumor microenvironment deemed unfavorable for immunotherapy.
As PIVOT-02 yielded strong synergistic effects, Bristol-Myers Squibb launched a phase 3 trial comparing NKTR-214 and nivolumab with nivolumab monotherapy [70]. The primary completion is estimated in 2024 and results have not yet been published.
6.2. Exploration of Novel Immunotherapeutic Targets: gp100
Glycoprotein (gp)100 (Figure 4) is a melanocyte-lineage specific antigen highly expressed in melanoma cells. Expressed gp100 in the context of HLA-A*0201 was seen to be detected and lysed by TILs [71]. However, the rate of recognition of self-derived antigens, as e.g., overexpressed in tumors, is low, due to a weak affinity of the T cell receptor (TCR) in order to reduce autoreactivity [72]. Immune-mobilizing monoclonal TCRs against cancer (ImmTACs) consist of a tumor-epitope-directed TCR and a CD3-specific, humanized single-chain antibody fragment and are thought to act as a link between tumor and T cells [73]. In melanoma, the domain of IMCgp100 (tebentafusp) exclusively binds to gp100 and might therefore redirect T cells to melanoma cells due to its CD3 antibody fragment.
Based on data of IMCgp100–202, a phase 2 trial, comparing tebentafusp with investigator’s choice (dacarbazine, ipilimumab, or pembrolizumab), the agent gained fast track designation for the treatment of metastatic uveal melanoma by the FDA [75]. In phase 1 of the IMCgp100–102 study, escalating doses of IMCgp100 were investigated in 19 heavily pre-treated patients with metastatic uveal melanoma, elevated LDH (87%) and liver metastases (100%) [76]. The 1-year OS rate was 74%. The IMCgp100-201 trial, a phase 1b/2 study, investigates tebentafusp alone or in combination with ICIs in patients with metastatic cutaneous melanoma [77]. The primary completion is estimated in 2022.
6.3. Adoptive T Cell Transfer
The idea of adoptive T cell transfer is to produce and select a large number of antitumor lymphocytes that recognize tumor cells with strong affinity. Moreover, inhibitory factors that are normally existing in vivo, are removed through in vitro activation [78]. One of the major limiting factors is the identification of cells selectively targeting tumor antigens and not the ones expressed on normal tissues [78].
The resected melanoma is split up into multiple tumor fragments individually grown in IL-2. Overgrowing lymphocytes lyse the tumor and pure cultures of lymphocytes with reactivity against the tumor arise. In around 5–6 weeks, it is possible to cultivate up to 1011 lymphocytes. An additional lymphodepletion appears to be able to increase the T cell persistence and the clinical response including the DOR [79].
At the ASCO 2020, data of C-144-01, a phase 2 trial including 66 patients with unresectable metastatic melanoma who have progressed on ICIs and BRAF/MEK inhibitors, were shown [80]. Tumor tissue was resected and TILs were produced ex vivo. The patients underwent nonmyeloablative lymphodepletion with cyclophosphamide followed by fludarabine; then they received one-time treatment of expanded and activated TILs and up to 6 doses of IL-2. The ORR was 36%, with 3% CR and 33% PR. The disease control rate was 80%; after a median follow up of 18.7 months, the median DOR has not been reached. Toxicity, most likely due to lymphodepletion and IL-2, was high, with grade 3 or 4 TRAEs in 97% of patients. Thrombocytopenia, anemia, and neutropenia, lasting a few weeks, were the most common AEs. One death was caused by intraabdominal hemorrhage and was considered to be possibly related to TILs. The other death was due to acute respiratory failure and not related to TILs per investigator assessment.
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65.
- Davies, M.A. The Role of the PI3K-AKT Pathway in Melanoma. Cancer J. 2012, 18, 142–147.
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis Primers 2015, 1, 15003.
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous Melanoma: From Pathogenesis to Therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080.
- Westphal, D.; Glitza Oliva, I.C.; Niessner, H. Molecular Insights into Melanoma Brain Metastases. Cancer 2017, 123, 2163–2175.
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844.
- Demkova, L.; Kucerova, L. Role of the HGF/c-MET Tyrosine Kinase Inhibitors in Metastasic Melanoma. Mol. Cancer 2018, 17, 26.
- Hartman, M.L.; Czyz, M. MITF in Melanoma: Mechanisms behind Its Expression and Activity. Cell Mol. Life Sci. 2015, 72, 1249–1260.
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master Regulator of Melanocyte Development and Melanoma Oncogene. Trends Mol. Med. 2006, 12, 406–414.
- Tsao, H.; Atkins, M.B.; Sober, A.J. Management of Cutaneous Melanoma. N. Engl. J. Med. 2004, 351, 998–1012.
- Agarwala, S.S. Current Systemic Therapy for Metastatic Melanoma. Expert Rev. Anticancer 2009, 9, 587–595.
- Allison, J.P.; Krummel, M.F. The Yin and Yang of T Cell Costimulation. Science 1995, 270, 932–933.
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J. Exp. Med. 1995, 182, 459–465.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020.
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U. ESMO Guidelines Committee. Electronic address: Cutaneous Melanoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1884–1901.
- Coit, D.G.; Thompson, J.A.; Albertini, M.R.; Barker, C.; Carson, W.E.; Contreras, C.; Daniels, G.A.; DiMaio, D.; Fields, R.C.; Fleming, M.D.; et al. Cutaneous Melanoma, Version 2.2019, NCCN Clinical Practice Guidelines in Oncol.ogy. J. Natl. Compr. Cancer Netw. 2019, 17, 367–402.
- Keilholz, U.; Ascierto, P.A.; Dummer, R.; Robert, C.; Lorigan, P.; van Akkooi, A.; Arance, A.; Blank, C.U.; Sileni, V.C.; Donia, M.; et al. ESMO Consensus Conference Recommendations on the Management of Metastatic Melanoma: Under the Auspices of the ESMO Guidelines Committee. Ann. Oncol. 2020, 31, 1435–1448.
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895.
- Ribas, A. Tumor Immunotherapy Directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519.
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma—An Update. J. Clin. Med. 2020, 9, 223.
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330.
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy. JAMA Oncol. 2019, 5, 187–194.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Previously Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546.
- Lebbé, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875.
- Hodi, F.S.; Kluger, H.; Sznol, M.; Carvajal, R.; Lawrence, D.; Atkins, M.; Powderly, J.; Sharfman, W.; Puzanov, I.; Smith, D.; et al. Abstract CT001: Durable, Long-Term Survival in Previously Treated Patients with Advanced Melanoma (MEL) Who Received Nivolumab (NIVO) Monotherapy in a Phase I Trial. Cancer Res. 2016, 76, CT001.
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results from an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol. 2019, 20, 1239–1251.
- Raedler, L.A. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96–100.
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Ӧhrling, K.; Kaufman, H.L. Final Analyses of OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec versus Granulocyte-Macrophage Colony-Stimulating Factor in Unresectable Stage III–IV Melanoma. J. Immunother. Cancer 2019, 7.
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516.
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39.
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636.
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and Trametinib versus Dabrafenib and Placebo for Val600 BRAF-Mutant Melanoma: A Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet 2015, 386, 444–451.
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876.
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib Combined with Vemurafenib in Advanced BRAF(V600)-Mutant Melanoma (CoBRIM): Updated Efficacy Results from a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2016, 17, 1248–1260.
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib in Patients with BRAF-Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615.
- Gogas, H.; Ascierto, P.A.; Flaherty, K.; Arance, A.; Mandalà, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Update on Overall Survival in COLUMBUS: A Randomized Phase III Trial of Encorafenib (ENCO) plus Binimetinib (BINI) versus Vemurafenib (VEM) or ENCO in Patients with BRAF V600-Mutant Melanoma. J. Clin. Oncol. 2020, 38, 10012.
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus Dacarbazine in Patients with Advanced NRAS-Mutant Melanoma (NEMO): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2017, 18, 435–445.
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, Vemurafenib, and Cobimetinib as First-Line Treatment for Unresectable Advanced BRAFV600 Mutation-Positive Melanoma (IMspire150): Primary Analysis of the Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2020, 395, 1835–1844.
- Nathan, P.; Dummer, R.; Long, G.V.; Ascierto, P.A.; Tawbi, H.A.; Robert, C.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; Mandala’, M.; et al. LBA43 Spartalizumab plus Dabrafenib and Trametinib (Sparta-DabTram) in Patients (Pts) with Previously Untreated BRAF V600–Mutant Unresectable or Metastatic Melanoma: Results from the Randomized Part 3 of the Phase III COMBI-i Trial. Ann. Oncol. 2020, 31, S1172.
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788.
- Liu, B.L.; Robinson, M.; Han, Z.-Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 Deleted Herpes Simplex Virus with Enhanced Oncol.ytic, Immune Stimulating, and Anti-Tumour Properties. Gene 2003, 10, 292–303.
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncol.ytic Viruses for the Treatment of Melanoma. Am. J. Clin. Derm. 2017, 18, 1–15.
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A Phase I Study of OncoVEXGM-CSF, a Second-Generation Oncol.ytic Herpes Simplex Virus Expressing Granulocyte Macrophage Colony-Stimulating Factor. Clin. Cancer Res. 2006, 12, 6737–6747.
- Andtbacka, R.H.I.; Agarwala, S.S.; Ollila, D.W.; Hallmeyer, S.; Milhem, M.; Amatruda, T.; Nemunaitis, J.J.; Harrington, K.J.; Chen, L.; Shilkrut, M.; et al. Cutaneous Head and Neck Melanoma in OPTiM, a Randomized Phase 3 Trial of Talimogene Laherparepvec versus Granulocyte-macrophage Colony-stimulating Factor for the Treatment of Unresected Stage IIIB/IIIC/IV Melanoma. Head Neck 2016, 38, 1752–1758.
- Andtbacka, R.H.I.; Ross, M.; Puzanov, I.; Milhem, M.; Collichio, F.; Delman, K.A.; Amatruda, T.; Zager, J.S.; Cranmer, L.; Hsueh, E.; et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann. Surg. Oncol. 2016, 23, 4169–4177.
- Long, G.V.; Dummer, R.; Ribas, A.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.S.; et al. Efficacy Analysis of MASTERKEY-265 Phase 1b Study of Talimogene Laherparepvec (T-VEC) and Pembrolizumab (Pembro) for Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 9568.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954.
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114.
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Invest. Derm. 2020.
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. The Mitogen-Activated Protein Kinase Pathway in Melanoma Part I–Activation and Primary Resistance Mechanisms to BRAF Inhibition. Eur. J. Cancer 2017, 73, 85–92.
- Kim, T.; Amaria, R.N.; Spencer, C.; Reuben, A.; Cooper, Z.A.; Wargo, J.A. Combining Targeted Therapy and Immune Checkpoint Inhibitors in the Treatment of Metastatic Melanoma. Cancer Biol. Med. 2014, 11, 237–246.
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory Effects of BRAF and MEK Inhibitors: Implications for Melanoma Therapy. Pharm. Res. 2018, 136, 151–159.
- Yu, C.; Liu, X.; Yang, J.; Zhang, M.; Jin, H.; Ma, X.; Shi, H. Combination of Immunotherapy With Targeted Therapy: Theory and Practice in Metastatic Melanoma. Front. Immunol. 2019, 10.
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Becker, J.C.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Livingstone, E.; Long, G.V.; et al. Survival of Patients with Advanced Metastatic Melanoma: The Impact of MAP Kinase Pathway Inhibition and Immune Checkpoint Inhibition-Update 2019. Eur. J. Cancer 2020, 130, 126–138.
- Reijers, I.L.M.; Rozeman, E.A.; Wilgenhof, S.; van Thienen, J.V.; Haanen, J.B.A.G.; Blank, C.U. Switch to Checkpoint Inhibition after Targeted Therapy at Time of Progression or during Ongoing Response: A Retrospective Single-Centre Experience in Patients with BRAF-Mutated Melanoma. Pigment. Cell Melanoma Res. 2020, 33, 498–506.
- National Cancer Institute (NCI). DREAMseq (Doublet, Randomized Evaluation in Advanced Melanoma Sequencing) a Phase III Trial; 2020. Available online: (accessed on 27 June 2020).
- Schadendorf, P.D. Med D. In A Phase II, Open-Label., Randomized-Controlled Trial Evaluating the Efficacy and Safety of a Sequencing Schedule of Cobimetinib Plus Vemurafenib Followed by Immunotherapy With an Anti- PD-L1 Antibody Atezolizumab for the Treatment in Patients With Unresectable or Metastatic BRAF V600 Mutant Melanoma; 2020. Available online: (accessed on 27 June 2020).
- Rozeman, E.A.; Versluis, J.M.; Sikorska, K.; Lacroix, R.; Grijpink-Ongering, L.G.; Heeres, B.; Van De Wiel, B.A.; Dimitriadis, P.; Sari, A.; Heijmink, S.; et al. The IMPemBra Trial, a Phase II Study Comparing Pembrolizumab with Intermittent/Short-term Dual MAPK Pathway Inhibition plus Pembrolizumab in Melanoma Patients Harboring the BRAFV600 Mutation. J. Clin. Oncol. 2020, 38, 10021.
- Arance, A.M.; Gogas, H.; Dreno, B.; Flaherty, K.T.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Ferrucci, P.F.; Pigozzo, J.; Rutkowski, P.; et al. Combination Treatment with Cobimetinib (C) and Atezolizumab (A) vs Pembrolizumab (P) in Previously Untreated Patients (Pts) with BRAFV600 Wild Type (Wt) Advanced Melanoma: Primary Analysis from the Phase III IMspire170 Trial. Ann. Oncol. 2019, 30, v906.
- Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. 2014, 192, 5451–5458.
- Dutcher, J.P.; Gaynor, E.R.; Boldt, D.H.; Doroshow, J.H.; Bar, M.H.; Sznol, M.; Mier, J.; Sparano, J.; Fisher, R.I.; Weiss, G. A Phase II Study of High-Dose Continuous Infusion Interleukin-2 with Lymphokine-Activated Killer Cells in Patients with Metastatic Melanoma. J. Clin. Oncol. 1991, 9, 641–648.
- Kruit, W.H.; Punt, C.J.; Goey, S.H.; de Mulder, P.H.; Gratama, J.W.; Eggermont, A.M.; Bolhuis, R.L.; Stoter, G. Dose Efficacy Study of Two Schedules of High-Dose Bolus Administration of Interleukin 2 and Interferon Alpha in Metastatic Melanoma. Br. J. Cancer 1996, 74, 951–955.
- Charych, D.H.; Hoch, U.; Langowski, J.L.; Lee, S.R.; Addepalli, M.K.; Kirk, P.B.; Sheng, D.; Liu, X.; Sims, P.W.; VanderVeen, L.A.; et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin. Cancer Res. 2016, 22, 680–690.
- Bentebibel, S.-E.; Hurwitz, M.E.; Bernatchez, C.; Haymaker, C.; Hudgens, C.W.; Kluger, H.M.; Tetzlaff, M.T.; Tagliaferri, M.A.; Zalevsky, J.; Hoch, U.; et al. A First-in-Human Study and Biomarker Analysis of NKTR-214, a Novel IL2Rβγ-Biased Cytokine, in Patients with Advanced or Metastatic Solid Tumors. Cancer Discov. 2019, 9, 711–721.
- Diab, A.; Tannir, N.M.; Bentebibel, S.-E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Efficacy, and Immune Activation (PIVOT-02). Cancer Discov. 2020.
- A Study of NKTR-214 Combined With Nivolumab vs Nivolumab Alone in Participants With Previously Untreated Inoperable or Metastatic Melanoma-Full Text View-ClinicalTrials.Gov. Available online: (accessed on 24 May 2020).
- Bakker, A.B.; Schreurs, M.W.; de Boer, A.J.; Kawakami, Y.; Rosenberg, S.A.; Adema, G.J.; Figdor, C.G. Melanocyte Lineage-Specific Antigen Gp100 Is Recognized by Melanoma-Derived Tumor-Infiltrating Lymphocytes. J. Exp. Med. 1994, 179, 1005–1009.
- Cole, D.K.; Pumphrey, N.J.; Boulter, J.M.; Sami, M.; Bell, J.I.; Gostick, E.; Price, D.A.; Gao, G.F.; Sewell, A.K.; Jakobsen, B.K. Human TCR-Binding Affinity Is Governed by MHC Class Restriction. J. Immunol. 2007, 178, 5727–5734.
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-Redirected Tumor Cell Killing. Nat. Med. 2012, 18, 980–987.
- Charych, D.; Khalili, S.; Dixit, V.; Kirk, P.; Chang, T.; Langowski, J.; Rubas, W.; Doberstein, S.K.; Eldon, M.; Hoch, U.; et al. Modeling the Receptor Pharmacology, Pharmacokinetics, and Pharmacodynamics of NKTR-214, a Kinetically-Controlled Interleukin-2 (IL2) Receptor Agonist for Cancer Immunotherapy. PLoS ONE 2017, 12, e0179431.
- Immunocore Ltd. A Phase II Randomized, Open-Label., Multi-Center Study of the Safety and Efficacy of IMCgp100 Compared With Investigator Choice in HLA-A*0201 Positive Patients With Previously Untreated Advanced Uveal Melanoma; 2020. Available online: (accessed on 27 June 2020).
- Sato, T.; Nathan, P.D.; Hernandez-Aya, L.; Sacco, J.J.; Orloff, M.M.; Visich, J.; Little, N.; Hulstine, A.-M.; Coughlin, C.M.; Carvajal, R.D. Redirected T Cell Lysis in Patients with Metastatic Uveal Melanoma with Gp100-Directed TCR IMCgp100: Overall Survival Findings. J. Clin. Oncol. 2018, 36, 9521.
- Phase 1b/2 Study of the Combination of IMCgp100 With Durvalumab and/or Tremelimumab in Cutaneous Melanoma-Full Text View-ClinicalTrials.Gov. Available online: (accessed on 24 May 2020).
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68.
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes. Science 2002, 298, 850–854.
- Sarnaik, A.; Khushalani, N.I.; Chesney, J.A.; Lewis, K.D.; Medina, T.M.; Kluger, H.M.; Thomas, S.S.; Domingo Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Long-Term Follow up of Lifileucel (LN-144) Cryopreserved Autologous Tumor Infiltrating Lymphocyte Therapy in Patients with Advanced Melanoma Progressed on Multiple Prior Therapies. J. Clin. Oncol. 2020, 38, 10006.

