 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Blanka Holendova | + 5499 word(s) | 5499 | 2021-04-27 09:36:53 | | | |
| 2 | Conner Chen | Meta information modification | 5499 | 2021-05-05 09:22:16 | | | | |
| 3 | Lydie Plecita-Hlavata | + 17 word(s) | 5516 | 2021-05-06 08:24:43 | | |
Video Upload Options
Redox status is a key determinant in the fate of every cell, β-cell in particular. β-cells are not primarily detoxifying like e.g. hepatocytes or kidney cells and thus do not possess extensive antioxidant defense machinery. However, they show a wide range of redox regulating proteins, such as peroxiredoxins, thioredoxins or thioredoxin reductases, etc., being functionally compartmentalized. These proteins keep fragile redox homeostasis and serve as messengers and amplifiers of redox signaling which is inevitable for proper β-cell function (particularly insulin secretion) and maintenance. Dysbalance in redox homeostasis establishes oxidative stress which accompanies the development of type 2 diabetes.
1. Redox Homeostasis in β-Cell Development and Maturation
1.1. Sources of Reactive Oxygen Species and Antioxidants in β-Cells
Reactive oxygen species (ROS) are normally produced during the metabolism of β-cells and play an important role in cellular signaling. For instance, mitochondrial ROS are obligatory signals of glucose-induced insulin secretion (GSIS) [1][2]. However, excessive levels of ROS in both human and rodent β-cells cause oxidative stress, which is detrimental to the cells. Several such conditions leading to excessive ROS generation in β-cells have been proposed, such as hyperglycemia, hypoxia, hyperlipidemia and endoplasmic reticulum (ER) stress [3]. Pancreatic β-cells both of rodents and humans are reportedly determined to be especially vulnerable to oxidative damage [4] due to the low expression of classical antioxidant enzymes—catalases, glutathione peroxidases (GPX) and superoxide dismutases (SOD) when compared to other cell types [5][6][7]. The main antioxidant system in β-cells consists of peroxiredoxins (PRX), thioredoxins (TRX) and thioredoxin reductase (TRXR). Regeneration of PRX thiol groups is mediated by auxiliary enzymes TRX and glutaredoxins (GRX). The recycling of TRX is mediated by the activity of TRXR, which reduces TRX and allows the cycle to continue. NADPH serves as an electron donor for the reduction of TRXR [8]. GRX is reduced by glutathione, which is then regenerated by glutathione reductase with the concurrent utilization of NADPH [8]. This system was shown to be sufficient to protect β-cells against short-run oxidative burst while hypothetically also providing a signaling role necessary for GSIS in both rodent and human cells [9]. Nevertheless, long-term glucolipotoxic conditions, often caused by overnutrition leading to an increase in oxidative stress, can cause dysfunction of the β-cells and contribute to type 2 diabetes. The redox balance between ROS and the antioxidant system is, therefore, crucial for the proper physiological function of pancreatic β-cells, and thus body glucose homeostasis. Because of the internal redox compartmentalization in β-cells, we have focused on the local sources of ROS and types of antioxidant enzymes in individual cell parts (Figure 1). ROS are primarily produced during mitochondrial oxidative phosphorylation but also originate from other cell compartments and organelles, such as ER, peroxisomes or cytoplasm [10][11]. At a “redox triangle” called redoxosome, which is formed by closely attached membranes of the ER, mitochondria and peroxisomes, redox-regulatory enzymes are thought to assemble. These sense ROS accumulations and redox imbalances, and its enzymes use ROS to transmit intercompartmental signals via chemical modifications of downstream proteins and lipids [12]. Under physiological conditions, ROS production in individual compartments is well controlled by specific resident antioxidant enzymes with small differences between rodents and humans.
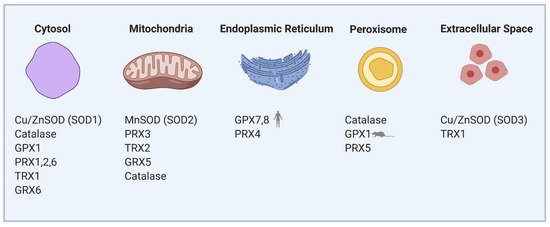
Figure 1. Compartmentalization of redox regulating proteins in β-cells involves mitochondria, cytosol, endoplasmic reticulum (ER), peroxisomes and extracellular space. SOD—superoxide dismutase, GPX—glutathione peroxidase, PRX—peroxiredoxin, TRX—thioredoxin, GRX—glutaredoxin.
1.1.1. Mitochondria
Mitochondrial electron transport chain (ETC) complexes I and III produce superoxide (O2•−) into the mitochondrial matrix and complex III also into the cytosol (exactly to the intracristal space). Mitochondrial MnSOD (Cu/ZnSOD isoform in the cytosol) transforms O2•− into a more stable molecule H2O2, which is then reduced to water by an antioxidant enzyme GPX, mitochondrial PRX3 or catalase [13]. Under physiological conditions in the presence of glucose, mitochondrial production of ROS is not detrimental to the β-cells. GSIS does not increase mitochondrial ROS production in rodent β-cells [2][14][15]. However, elevated H2O2 derived from superoxide formed by fatty acid oxidation (specifically by electron transfer flavoprotein-ubiquinone oxidoreductase) activates mitochondrial phospholipase iPLA2γ. iPLA2γ-cleaved fatty acids are being utilized by UCP2 to induce mild uncoupling and subsequently reduce respiratory chain-mediated superoxide formation. Such an antioxidant feedback mechanism based on redox signaling initiated by fatty acid oxidation in rodent β-cells might suppress further ROS production and oxidative stress. Also, iPLA2γ-cleaved fatty acids were shown to amplify insulin secretion in mice through the GPR40 receptor [16][17]. This way, mitochondria may help the cells to cope with the nutrient overload.
1.1.2. Endoplasmic Reticulum
During the maturation of insulin in the ER, three disulfide bonds are formed by disulfide isomerase (PDI). After the formation of the S–S bond, PDI needs to be regenerated to its oxidized form. This is enabled either by ER oxidoreductin-1 (ERO1), which accepts electrons from PDI and donates them to oxygen resulting in the formation of H2O2 molecules or by GSSG-driven oxidation of substrate proteins through PDI (Figure 2). The interplay between the two oxidative pathways that produce or consume ER-luminal GSSG maintains ER redox status (manifested in human HEK293 cell line) [18]. H2O2 is then metabolized by PRX4 in both rodents and humans, together with GPX7/8 in humans [19][20][21][22]. Overexpression of PRX4 in rat INS-1E cells protects against ROS toxicity and increases insulin content by affecting ER protein folding capacity [19]. Redox balance on the ER level affects other membranes of the redoxosome. Long-term overproduction of ROS in ER leads to cell stress, activation of unfolded protein response, β-cell dedifferentiation and subsequently apoptosis. The state of protein folding in ER is controlled by chaperones of the Hsp70 and Hsp40 families, chaperone-like proteins, and lectins [23][24]. The key ER members of the Hsp70 family are represented by chaperone immunoglobulin heavy-chain-binding protein, BiP (also called GRP78) [25]. UPR signaling is mediated via three main transmembrane sensors: endoribonuclease inositol-requiring protein 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) [26]. When misfolded proteins accumulate in the ER, BiP dissociates from the UPR sensors and binds to the exposed hydrophobic domains of the unfolded proteins [27]. This induces oligomerization and auto-transphosphorylation of IRE1α and PERK, with their consequent activation [28].
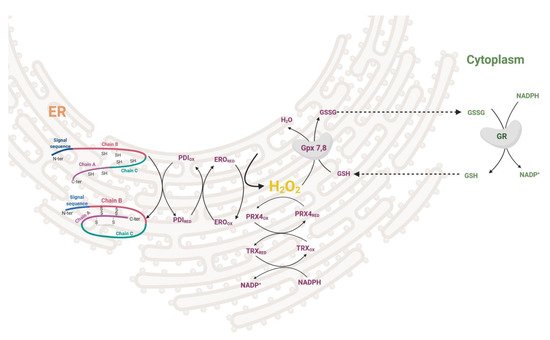
Figure 2. Redox regulation in ER during proinsulin folding. Disulfide bonds are formed by disulfide isomerase (PDI) together with ER oxidoreductin (ERO) oxidoreductases while giving rise to H2O2. This can be attenuated by PRX4/TRX or GPX7,8/GSH redox couples in conjunction with cytosolic redox status. PDI—disulfide isomerase, ERO—ER oxidoreductin, PRX—peroxiredoxin, TRX—thioredoxin, GPX—glutathione peroxidase, GSH—glutathione.
1.1.3. Peroxisomes
In peroxisomes, ROS are produced mainly during the β-oxidation of long-chain polyunsaturated fatty acids. Because of the weak antioxidant defense of peroxisomes in both rodents and humans, β-cells are unable to endure long-term peroxisomal stress. Catalase expression is rather suppressed to allow specific β-cell function [5][6]. Also, Prx5 is weakly expressed [20]. Thus, weak detoxifying machinery in peroxisomes leads to uncontrollable ROS production in lipotoxic conditions. However, the peroxisomal loss causes mitochondrial deterioration and cytoplasmic vacuolization in β-cells impairing glucose tolerance [29].
1.1.4. Cytoplasm and Membrane Rafts
In our previous research on mice [2], we have shown that after glucose uptake, the ROS production arises mainly in the cytoplasm, where specific H2O2 producers are localized: NADPH oxidases (NOX) and xanthine oxidases (XOD) [30][31][32]. These enzymes have an important role in the physiology but also the pathophysiology of pancreatic β-cells. Under physiological conditions, the cytoplasmic antioxidant system is fully sufficient to maintain the redox balance. During GSIS, the levels of NADPH and mitochondrial FADH2 are increased, NAD(P)H-dependent ROS production is stimulated, and detoxifying enzymes (NOX, XOD and PRX/TRX/TRXR system, respectively) are activated. The primary cytoplasmic antioxidants are PRX1, PRX2 [33] and, to a lower extent, also Cu/ZnSOD, catalase and GPX1 [5][6][11][20] (Figure 1). Thioredoxin interacting protein (TXNIP) was suggested to increase oxidative stress by binding TRX and inhibiting its reducing activity [34]. Upregulated by glucose, TXNIP regulates β-cell antioxidant defense and redox homeostasis. Carbohydrate response element-binding protein (ChREBP) participates in TXNIP expressional regulation, thus linking redox balance to glucose sensing through the ChREBP–TXNIP–TRX axis both in humans and rodent β-cells [35]. Besides TRX, many other proteins were suggested to be the binding targets to the TXNIP protein; thus, TXNIP may be a scaffold protein important in redox signaling [36]. As discovered on human cancer cell line MCF-7, one of TXNIP’s binding partners is importin-α, which allows the complex to directly enter the nucleus [37]. TXNIP overproduction during long-time hyperglycemia has a deleterious effect on β-cell function also through controlling microRNA (miR) expression. Through upregulation of miR-204, TXNIP downregulates expression of V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), one of the major insulin transcription factors [38]. Induction of another miR-124a expression elevates the production of islet amyloid polypeptide, further aggravating the Langerhans islets pathology [39].
1.2. ROS Signaling Pathways and Transcription Factors in β-Cell Physiology
The exact mechanisms by which ROS (mainly H2O2) serve as signaling molecules and metabolic coupling factors in well-functioning β-cells are still not fully understood. However, there is strong evidence of ROS importance in proper β-cell function [40]. It was found that administration of exogenous H2O2 and enhancing intracellular H2O2 levels increases insulin release in rat INS-1 (832/13) cells and both rat and mouse islets [41][42][43]. On the contrary, the application of exogenous antioxidants inhibits GSIS [43]. How could H2O2 (or other ROS) mediate its signaling effects with the antioxidant system present in the cell? In general, three main mechanisms of H2O2 function have been proposed. First, there may be strong localized intracellular H2O2 gradients that enable local H2O2 signaling despite the mean cytoplasmic concentration being low. Second, PRX may be locally transiently inactivated during signaling, allowing H2O2 levels to increase. Third, the action of H2O2 may be mediated by redox relays whereby PRX and/or glutathione peroxidases and catalase to less extent act as initial H2O2 sensors and then oxidize the end target proteins [44]. Redox sensing and signaling within the cells are mediated via reversible disulfide-dithiol exchange reactions and de-nitrosylation of Cys residues. The process is enzymatically controlled by members of the thioredoxin family comprising over 50 proteins, including TRX, PRX, PDI, glutathione-dependent glutaredoxins (GRX), GPX and glutathione-transferases [45]. The enzymes interact with many distinct proteins affecting cellular functions like gene expression [46], regulation of intracellular H2O2 levels [33], proliferation [47], apoptosis [48] and modulation of the immune response [49].
1.2.1. Redox Sensitive Signaling Pathways
Several cellular pathways were shown to be affected by redox status in β-cells. Signaling pathway KEAP1–NRF2–ARE, in general, maintains the cellular redox balance and induces adaptive response to oxidative stress [50]. This pathway consists of three main components: Kelch-like ECH-associated protein 1 (KEAP1), nuclear factor erythroid 2-related factor 2 (NRF2), and antioxidant response elements (ARE). Under physiological conditions, KEAP1 is associated with NRF2, and the ubiquitination of NRF2 is stimulated. However, under oxidizing conditions, ROS promote the dissociation of KEAP1 and NRF2, enabling NRF2 to transfer to the nucleus, where it binds to ARE and enhances the expression of detoxification enzymes, such as glutathione synthetase, glutathione reductase, GPX, TRX, TRXR and PRX to prevent oxidative stress [50]. Elevated NRF2 activity has a positive effect on β-cell function during glucolipotoxicity in mice [51]. However, under physiological conditions, insulin release is reduced after activation of NRF2 [51]. Chronic H2O2 treatment is known to activate KEAP1–NRF2–ARE signaling to promote the production of pro-apoptotic factors and leads to apoptosis of human pancreatic β-cells [52]. ROS were also shown to activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. NF-κB, when activated, translocates to the nucleus and works as a transcription factor that affects many cellular processes. This pathway can influence the redox status by increasing the expression of antioxidant proteins, such as Cu/ZnSOD, MnSOD, GPX, etc. [50]. However, NF-κB activation in pancreatic β-cells has mostly deleterious effects and contributes to the loss of differentiated β-cell functions and leads to cell death [53][54][55]. Another major intracellular signal transduction pathway that plays an important role in various cellular processes is the mitogen-activated protein kinase (MAPK) cascades. This includes the extracellular signal-regulated kinase (ERK) pathway, the c-Jun N-terminal kinase (JNK) pathway, the p38 kinase pathway and the big MAP kinase 1 (BMK1/ERK5) pathway. ROS were reported to activate these pathways under physiological and pathophysiological conditions of β-cells [50][56].
1.2.2. Redox Regulation of Transcription Factors Essential for β-Cell Maturation
For a long time, activation of gene transcription has been considered to be primarily, if not exclusively, regulated by cascades of protein phosphorylation and dephosphorylation [57]. The concept that transcription might be controlled by redox reactions emerged when an important eukaryotic transcription factor–NF-κB–was found to be activated by oxidants and inhibited by antioxidants [57][58][59]. Several theories how oxidants could affect transcription were suggested—oxidative inactivation of phosphatases in signaling cascades, direct oxidation of transcription factors, activation of protein kinases, redox-dependent noncovalent binding of thioredoxin, thiol modifications of proteins that form cytosolic complexes with transcription factors, or heterodimer formation of GPX and PRX-type peroxidases with transcription factors. In the mature pancreas, transcription factors play a role in achieving glucose homeostasis by regulating the expression of key genes, most notably the insulin gene [60]. Mature β-cells of all species express high levels of transcriptional factors. The main ones include Pancreas/duodenum homeobox protein 1 (PDX1), MAFA, homeobox protein NKX6.1 and transcription factor neurogenic differentiation (NEUROD). All of these transcription factors were shown to be downregulated by persistent ROS in rodents and human β-cells [3]. However, these changes occur under pathophysiological conditions of oxidative stress and often lead to cell damage. PDX1 is essential in the development of β-cells and also binds to the regulatory elements and increases insulin gene transcription and several other islet-specific genes like glucose transporter2 (Glut2) or Nkx6.1 [60]. It contributes to β-cell mass expansion and glucose metabolism induced by activation of protein kinase B (PKB/Akt) signaling [61]. The activity of PDX1 was shown to be reduced by oxidative stress [62][63]. Another critically important activator of insulin gene transcription, MAFA, is sensitive to proteasomal degradation under conditions of oxidative stress in rodents [64][65]. The major regulator of MAFA degradation under oxidative stress is p38 MAPK, which directly binds to MAFA and triggers MAFA degradation via the ubiquitin proteasomal pathway. Inhibiting MAFA degradation under oxidative stress in rodents ameliorates β-cell dysfunction [66][67]. Reduction of intranuclear MAFA levels was observed in diabetic db/db mice. This adverse effect was prevented by β-cell-specific GPX1 overexpression, which induced β-cell antioxidant protection [68]. Both of the above-mentioned transcription factors are regulated by forkhead box O protein 1 (FOXO1) (). FOXO1 belongs to the FOXO family, which has an important role in many cellular processes, including resistance to cellular oxidative stress itself, in cellular metabolism and insulin signaling [69]. FOXO can sense ROS levels by oxidation of its cysteines and then regulates the activity of transcription factors. In humans, ROS induces the formation of cysteine-thiol disulfide-dependent complexes of FOXO and the p300/cAMP-responsive element-binding protein (CREB) binding protein (CBP) acetyltransferase, thus modulating FOXO biological activity [70]. Multiple FOXO connected pathways have been shown to be subject to regulatory cysteine oxidation [71]. The insulin and insulin-like growth factor (IGF) signaling pathways inhibit FOXO activity by activation of phosphoinositide 3-kinase (PI3K) and subsequently PKB [72] (Figure 3). Studies performed mainly on rodents showed that phosphorylation of FOXO by PKB causes FOXO inactivation and its export from the nucleus [69][71]. Under oxidative stress, H2O2-induced oxidation of PKB leads to the transformation of its Cys297 and Cys311 into intramolecular disulfide bonds [73][74], dephosphorylation of PKB and an increased association with protein phosphatase 2A (PP2A) (Figure 3). The higher affinity of PKB to PP2A leads to faster inactivation of this kinase, which enables nuclear localization of FOXO [71]. Interestingly, activated FOXO1 negatively regulates Pdx1 expression during oxidative stress in βTC-3 cell culture and mice [75]. It was also discovered that under oxidative burst, FOXO1 can bind to promyelocytic leukemia protein (PML) and NAD-dependent deacetylase sirtuin-1 (SIRT1) forming a single complex with a slightly inverse effect. This complex upregulates Ins2 gene transcription factors MAFA and NEUROD expression [76]. MAFA has an important role in β-cell development and replication. Shift from MAFB to MAFA expression during early development in β-cells has a significant role in β-cell maturity determination, replication and survival (reported on rodent model) [77]. This way FOXO1 in the short-term protects β-cells against oxidative stress and tries to preserve their proper function [3][76]. More investigations have to be made to clarify this dual effect of FOXO on β-cells. Apoptosis signal-regulating kinase 1 (ASK-1) is also activated in a redox-dependent manner. ASK-1 activates JNK, which leads to phosphorylation of FOXO and prevention of its export from nucleus [71]. Family of paired-homeobox genes (PAX) proteins (paired-homeobox genes) being crucial for islet development also occurs to be sensitive to redox regulations. PAX8 loses its ability to bind DNA molecules after oxidation of its cysteines Cys45 or Cys57. These cysteines are conserved in all Pax members, so the redox regulation occurs also in human and rodent β-cells, where PAX4 and PAX6 are present [78][79][80].
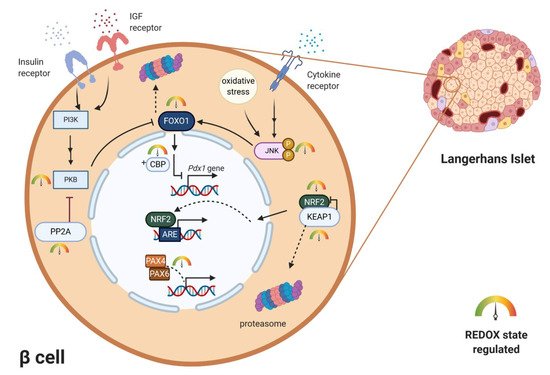
Figure 3. Examples of redox regulation of specific transcription factors. Pdx1 gene is negatively regulated by redox-sensitive transcription factors FOXO and CBP. Localization and transcription activity of FOXO is negatively regulated by PI3K-PKB/AKT signaling and positively regulated by the JNK signaling pathway. Activated by oxidative stress/cytokine stimuli, JNK enables FOXO nuclear localization. Concurrently redox-sensitive PKB is inhibited by H2O2-induced intramolecular disulfide bond formation. Redox sensitive NRF2/KEAP1 system regulates expression of key antioxidant enzymes upon ARE region. Under oxidizing conditions, the cytoplasmic complex dissociates, NRF2 enters the nucleus and functions as a transcription factor. Redox-sensitive cysteines in PAX transcription factors directly regulate their DNA-binding ability. ARE—antioxidant response elements, CBP—p300/cAMP-responsive element-binding protein (CREB)-binding protein, FOXO—forkhead box O protein, IGF—insulin-like growth factor, JNK—c-Jun N-terminal kinase, KEAP1—Kelch-like ECH-associated protein 1, NRF2—nuclear factor erythroid 2-related factor 2, PAX4/6—paired-homeobox genes, PDX1—pancreas/duodenum homeobox protein 1, PI3K—phosphoinositide 3-kinase, PP2A—protein phosphatase 2A, PKB—protein kinase B.
1.3. Redox Regulation of β-Cell Differentiation and Proliferation
1.3.1. Redox Regulation of β-Cell Differentiation
During differentiation of β-cells from embryonic stem cells, strictly controlled repression or activation of specific transcription factors and genes that manage the transition occurs. During the embryonic days 8.5–12.5, expression of specific transcription factors is activated: Sox17, Pdx1, Hb9, Isl1, Hnf1β, Foxa2, Hnf6, Gata4 and Gata6. Between embryonic days 12.5–16.5, other transcription factors are activated: Ngn3, NeuroD1, Pax4, Pax6, Hes1, Hnf6, Sox9, Ptf1a, Rfx3, Rfx6, Isl1, Arx, Nkx2.2, Nkx6.1, Glis3, MafA and MafB [81][82][83][84]. Mutations of these genes and their deficiency during early embryonic days lead to reduced β-cell mass, hyperglycemia and development of diabetes mellitus in rodents [84]. Moreover, new approaches in the treatment of diabetes rely on the conversion of differentiated adult cells or progenitors into β-cells in a process called cellular reprogramming and the promotion of their further proliferation [85]. Identifying and understanding key regulators specific for the development and identity of β-cells is, therefore, necessary. Redox signaling was suggested to be one of these regulators as treatment of pancreatic progenitors by low ROS levels leads to β-cell formation and its viability increase [86][87]. In murine embryos, H2O2 production positively correlates with β-cell formation. Moreover, NOX4 production of H2O2 stimulates the differentiation of β-cells progenitors accompanied by positive effects on Sox9 and Ngn3 gene expression in vivo and in vitro. Low levels of H2O2 promote the expression of differentiation markers Ngn3 and Nkx6.1 and markers of maturation [88]. On the contrary, reducing H2O2 concentration leads to a decrease in β-cell development, viability and proliferation [86][89] and overexpression of catalase, specifically in mice mitochondria of β-cells leads to a significant reduction in β-cell proliferation and volume [87]. β-cell differentiation is mediated probably through modulation of the ERK1/2 signaling pathway, which controls proliferation, differentiation and also phosphorylation and protein levels of CREB [89][90]. CREB, together with its coactivator CBP participates in β-cell gene expression and proliferation. In mutated mice and isolated islet cultures, constitutively activated cAMP signaling and enhanced CREB–CBP binding leads to an increase in proliferation and doubling β-cell mass [91]. The human SOX9 proximal promoter is also regulated by the CREB in other cell types [92]; thus, the positive effect of H2O2 from NOX4 on Sox9 and Ngn3 genes could be connected to the activation of the ERK1/2 signaling. In other cell types, the influence of H2O2 on the activation of signaling pathways connected to β-cell proliferation was discovered: PI3K/PKB pathway [93] and WNT signaling pathway [94].
1.3.2. Adipokines Stimulate β-Cells Proliferation through Redox Signaling
Hormones leptin and adiponectin are secreted by adipose tissue and play a major role in the homeostasis of the organism and β-cell condition. They control body weight, food intake and overall metabolism [95][96]. Many studies have been performed to elucidate the effects of leptin and adiponectin on β-cells; however, results have been controversial [96]. There is a negative feedback between insulin and leptin secretion. Insulin promotes leptin release, and leptin then inhibits insulin production and secretion [97] by opening the KATP sensitive channels [98] and inhibiting protein phosphatase 1 [99], important in vesicle docking and exocytosis (well discussed in [100]). The effect of adiponectin on insulin secretion seems to rely on glucose concentration. Studies on rodent and human models showed that adiponectin does not affect basal insulin secretion; however, it stimulates insulin secretion at high glucose [96][101]. Adiponectin improves insulin sensitivity in target tissues due to the suppressive effect on gluconeogenesis and positive influence on glucose uptake and fatty acid oxidation in muscle and liver [96]. However, the exposition of β-cells to elevated levels of these adipokines leads to a significant increase in their proliferation and viability. This is due to the increase in NOX activity and thus an increase in ROS production, which was confirmed by using exogenous antioxidants. The positive effect of adipokines on ROS elevation is accompanied and supported by a decrease in the Cu/ZnSOD expression and Cu/ZnSOD, GPX and catalase activity in rodents [86].
1.3.3. ROS-Mediated Ca2+ Signaling in β-Cell Proliferation
Due to a direct effect of ROS on redox-sensitive Ca2+ channels [11][102], ROS-mediated intracellular rise in Ca2+ concentration can lead to activation of calcineurin, which potentiates both human and rodent β-cell proliferation [103][104]. Calcineurin phosphorylation of the Transducer of regulated CREB activity-2 (TORC2) leads to its translocation into the nucleus and association with other transcription factors (CREB, cAMP-responsive element modulator (CREM) and cyclic AMP-dependent transcription factor ATF-1 (ATF1)) leading to enhanced expression of cell-cycle-activating genes [103]. At low nutrient levels, when ROS production and Ca2+ signaling in β-cells are generally low, AMPK kinase member microtubule affinity regulating kinase 2 (MARK2) is activated. MARK2 has an opposite effect on TORC2 than calcineurin; thus, starvation blocks β-cell proliferation [103].
2. Redox Homeostasis in Optimum β-Cell Function
2.1. Redox Signaling in Insulin Secretory Pathway
β-cells can adapt the rate of insulin secretion to the plasma concentration of glucose and other nutrients by a unique coupling system between nutrient metabolism and insulin production. The coupling system involves accelerating glycolysis, and mitochondrial metabolism with rapid changes in the oxidation state of several redox couples, e.g., NADH/NAD+ and NADPH/NADP+ and is also directly linked to ROS production as mentioned above. Upon higher substrate load, the demand for oxygen supply to the cells grows as well. At such a high metabolic activity, the oxygen demand exceeds oxygen supply, resulting in the establishment of intracellular transient hypoxia [105]. Such features have been described, for example, in neurosecretory cells, which require high mitochondrial activity and ATP production to restore resting membrane potential and to maintain intracellular Ca2+ levels [106] and also in working skeletal muscle cells, in which increased ATP demand is reflected by mitochondrial biogenesis and elevated oxygen consumption resulting in expression of hypoxia-inducible genes [107]. Hypoxia could serve as another coupling factor via enhanced ROS production for insulin secretion in β-cells. Moreover, the beneficial effect of hypoxia in healthy β-cells could be attributed to the biogenesis of the mitochondrial respiratory chain proteins [107] and effective calcium signaling [106], which are inevitable for the maintenance of mature β-cells. It was also suggested that the basal activity of hypoxia-induced pathways is necessary for normal β-cell function [108]. An interesting study by Olsson and Carlsson [109] has shown that oxygenation may differ widely between individual islets within the pancreas at a given time point. These differences may reflect a mechanism to recruit only a fraction of the available islets into an active (normoxic) β-cell mass. The remaining less well-oxygenated (hypoxic) islets may represent a dormant subpopulation, constituting a functional reserve of endocrine cells. According to this model, the reserve islet pool may be available for recruitment upon reduction of the total islet mass [109]. In any case, the importance of redox signaling in GSIS machinery was also documented by our research group, which showed recently that insulin secretion is inhibited upon deletion of constitutively active NOX4 enzyme in rodent β-cells in vivo. Our results demonstrated that the sole increase in ATP/ADP ratio is not sufficient to stimulate insulin secretion in vitro and in vivo. We thus confirmed that the presence of ROS, namely H2O2 as a metabolic coupling factor, is essential for insulin secretion [2]. Many targets of redox signaling in the insulin secretory pathway were suggested (Figure 4); however, a more detailed/complex view is still missing. KATP channel with its essential function in GSIS [110] mediating depolarization of plasma membrane was shown to be inhibited by H2O2 in smooth muscle cells [111], but no such regulation has been observed in β-cells. Also, another depolarizing transient receptor potential (TRP) channel subfamily M member (TRPM2) was reported to be directly redox activated [112]. The subsequent repolarizing events on the plasma membrane seem to be potentially redox-regulated as H2O2 could directly or indirectly inhibit repolarizing K+-channels, such as KV [113][114][115]. Besides redox targets of signaling pathways mentioned above, downstream aims in the secretory pathway were found to be, for example, redox-sensitive proteins of calcium signaling, e.g., ryanodine receptor 2 (RyR2), sarcoplasmic Ca2+ ATPase (SERCA) and inositol triphosphate receptor (IP3R) [11][102]. The glucose-induced changes in the redox environment affect these proteins, leading to increased exocytosis of insulin secretory vesicles. Further, proteins of the secretory machinery were also implicated to be redox-regulated. For example, redox-regulated posttranslational modification of exocytosis-regulating t-SNARE proteins has been proposed to result in an increased rate of maturation, or priming, of secretory vesicles in yeasts (for an overview, see [116]). mRNA/protein expression studies and electrophysiological analysis of exocytosis suggested that both the expression level of Grx1 and Trx1 in the β-cell as well as the NADPH/NADP+ redox status are important factors for the regulation of exocytosis [117]. Redox-dependent regulation was suggested for the NADPH–GSH–GRX1 signaling axis in rodent β-cells [118], and GRX1-GSH was shown to mediate deSUMOylation of sentrin/SUMO-specific protease 1 (SENP1) in humans [119] via modulation of its key thiol groups [120]. SUMOylation/deSUMOylation processes were previously reported to be redox-regulated and were implicated to be involved in response to oxidative stress [121]. Moreover, SUMOylation of insulin secretory pathway proteins had an inhibitory effect on insulin granule exocytosis, and several proteins involved in insulin granule exocytosis were shown to be modified by SUMOylation [121]. However, it is tempting to speculate that the redox signal mediated by the thiol exchange is transformed at the exocytotic site to signal mediated via SUMO posttranslational modification.
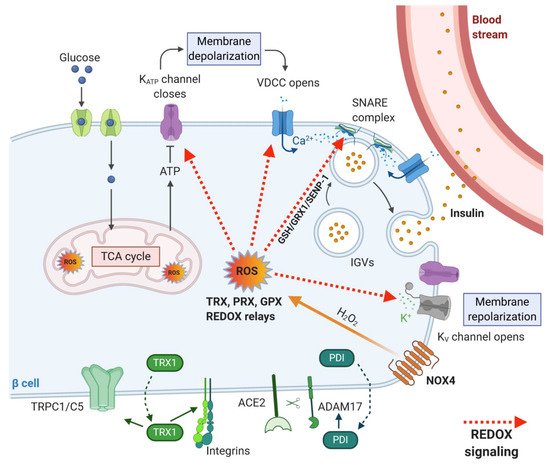
Figure 4. Redox targets within insulin secretory pathway and proteins involved in extracellular redox signaling. Glucose entry through GLUT1 in humans or GLUT2 in rodents causes an increase in ATP/ADP ratio and elevation of ROS production in the cytoplasm (mediated by NOX4). Subsequently, the KATP channel closes, which causes depolarization of the plasma membrane, the opening of voltage-dependent calcium channels (VDCC) and Ca2+ entry into the cells. Ca2+ is required for insulin granule vesicles (IGV) trafficking, attachment and fusion with the plasma membrane (mediated by SNARE proteins) and finally, secretion of insulin from the cell. Repolarization of the membrane is mediated via voltage-gated K+ channels (KV). These repeating cycles lead to a high-frequency of action potential spikes and synchronized oscillations in the cytosolic Ca2+. ROS are utilized by redox-regulated proteins TRX, GPX and PRX, which can either neutralize the ROS action in the long-term or transform acute redox signal via redox relays. Targets of redox signaling involve KATP channels, VDCC, KV channels and proteins involved in granule exocytosis. TRX1 and PDIs are secreted from β-cells, and they interact with extracellular domains of transmembrane proteins. PDIs regulate the action of sheddase ADAM17, and TRX1 modifies the activity of integrins and TRPC1/5 channels.
2.2. Extracellular Redox Signaling in β-Cell Function
It was shown that many oxidoreductases could be secreted from the cells to the extracellular milieu as a response to changes in the cellular environment [122]. The involved secretory pathways differ based on secreted enzyme and cell type. GRX1 is secreted by human T-cells, B-cells and NK-cells of the immune system [123]. Different PDIs are secreted by platelets playing a crucial role in thrombosis [124]. PRXs were detected to be secreted by macrophages and HEK297 cells [125]. TRX1 is secreted by many different cell lines, including human fibroblasts, epithelial cells, cells of the immune system, hepatocytes and cancer cells [126]. Interestingly, current research revealed secretion of cytoplasmic isoforms of TRX1 and TRXR1 by murine and porcine pancreatic β-cells during hypoxia and inflammation [126]. Such phenomenon was originally described in immune cells and presence of TRX1 was detected in blood of patients suffering from rheumatoid arthritis [127] and diabetes [128]. Extracellular TRX1 seems to cause autocrine or paracrine regulation of the mouse β-cells as recombinant TRX1 injected to mice with transplanted pancreatic islets significantly improved cell viability and improved blood glucose control, while overexpression of TRX1 in grafted islet cells did not lead to such effect [129]. In other experiments extracellular TRX1 had beneficial effect on murine and porcine β-cells by preventing apoptosis and preserving insulin secretion [126]. It is also possible that binding of TXNIP to TRX1 could negatively affect its secretion and subsequent extracellular actions. It is highly likely that except from TRX1 β-cells secrete also other oxidoreductases and that all these enzymes contribute to extracellular redox signaling towards other endocrine cells. More experimental evidence in this field will definitely reveal remarkable findings in future.
Until now, there is only a very limited number of transmembrane proteins, which were described as targets of extracellular redox modifications by secreted oxidoreductases. PDIs were pointed out as inactivators of ADAM metallopeptidase domain 17 (ADAM17) shedding activity [130][131][132]. ADAM17 is a disintegrin and metalloproteinase that participates in the cleavage of surface proteins. An increase in ADAM17-mediated shedding activity and decrease in its substrate angiotensin-converting enzyme 2 (ACE2) have been observed with the progression of type 2 diabetes mellitus (T2D) [133], suggesting the importance of this mechanism in the disease. Indeed, restoration of ACE2 improved glycemia in db/db and Ang-II-infused mice. The beneficial effects of ACE2 could be attributed to reduced oxidative stress and ADAM17 expression in the islets of Langerhans, in addition to the improvement of blood flow to the β-cells [134]. It seems probable that the regulation mediated by the PDIs–ADAM17–ACE2 axis may play an important role in sensing the redox environment in the β-cells and their surrounding, resulting in signaling events leading to maintenance of the proper β-cell functionality.
Another group of surface proteins described as targets of TRX1 and PDIA6 are the integrins [135][136]. Integrins are transmembrane heterodimer receptors for extracellular matrix (ECM) components. They are expressed on all cell types and composed of one α and one β subunit. Integrins affect a variety of cell processes, including adhesion, migration, differentiation, and cell growth. In mammals, 24 α and 9 β subunits have been identified, which combine to create 24 unique heterodimers [137]. Both the α and β subunits are involved in the determination of ligand specificity; however, the β subunit facilitates adhesion and activates intracellular second messenger cascades [138]. Most notably, stimulation of the β1 integrin leads to activation of focal adhesion kinase (FAK), ERK, PKB, and the steroid receptor coactivator (SRC) family of kinases, all of which are involved in cell survival. Moreover, integrins also mediate crosstalk with growth factor receptors. The signals that are activated by the integrin–FAK–SRC complex can be integrated with signals from growth factor receptor binding to subsequently activate the RAS–MAPK kinase–MAPK pathway, which is canonically associated with cell proliferation and survival [139]. Ligands for integrins include fibronectin, laminin, collagen, fibrinogen, and thrombospondin [137]. Regulation of disulfide formation and cleavage by TRX1 and PDIs could, therefore, affect β-cell growth, proliferation and survival- processes mediated by integrins.
The last group of redox-regulated transmembrane proteins that must not be forgotten are TRP channels. They comprise six divergent subfamilies TRPV, TRPC, TRPM, TRPA1, TRPP and TRPML, with diverse biophysical properties and functions. The TRP channels have a common structure of homo- or heterotetramers with six transmembrane spanning regions and intracellular N- and C-termini. TRP channels are sensitive to changes in the microenvironment, including heat, osmotic pressure, mechanical force and changes in the redox environment of the cells. They were found to be sensitive to redox modifications (for an overview, see [112]). It has been reported that the extracellular reduced thioredoxin activates homomeric TRPC5 and heteromeric TRPC5–TRPC1 channels by breaking a disulfide bridge in the predicted extracellular loop next to the ion-selectivity filter of TRPC5 [140]. It has been shown that TRPC5 and TRPC1 are expressed in secretory fibroblast-like synoviocytes from patients with rheumatoid arthritis, whose extracellular concentration of thioredoxin is high. The endogenous TRPC5–TRPC1 channels of the cells are activated by reduced thioredoxin, and the blockade of the channels enhances the secretory activity and prevents the suppression of secretion by thioredoxin. As patients with diabetes also have high levels of extracellular TRX1, one can predict that a similar mechanism also exists in pancreatic β-cells. It was also documented that activation of multiple subtypes of TRP channels results in enhanced insulin production by β-cells, and redox signal causes activation of the channels [141].
References
- Leloup, C.; Tourrel-Cuzin, C.; Magnan, C.; Karaca, M.; Castel, J.; Carneiro, L.; Colombani, A.L.; Ktorza, A.; Casteilla, L.; Penicaud, L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 2009, 58, 673–681.
- Plecita-Hlavata, L.; Jaburek, M.; Holendova, B.; Tauber, J.; Pavluch, V.; Berkova, Z.; Cahova, M.; Schroeder, K.; Brandes, R.P.; Siemen, D.; et al. Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes 2020.
- Wang, J.; Wang, H. Oxidative Stress in Pancreatic Beta Cell Regeneration. Oxid. Med. Cell. Longev. 2017, 2017, 1930261.
- Grankvist, K.; Marklund, S.L.; Taljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398.
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466.
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742.
- Miki, A.; Ricordi, C.; Sakuma, Y.; Yamamoto, T.; Misawa, R.; Mita, A.; Molano, R.D.; Vaziri, N.D.; Pileggi, A.; Ichii, H. Divergent antioxidant capacity of human islet cell subsets: A potential cause of beta-cell vulnerability in diabetes and islet transplantation. PLoS ONE 2018, 13, e0196570.
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry 2008, 73, 1493–1510.
- Stancill, J.S.; Broniowska, K.A.; Oleson, B.J.; Naatz, A.; Corbett, J.A. Pancreatic beta-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J. Biol. Chem. 2019, 294, 4843–4853.
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180.
- Roma, L.P.; Jonas, J.C. Nutrient Metabolism, Subcellular Redox State, and Oxidative Stress in Pancreatic Islets and beta-Cells. J. Mol. Biol. 2019.
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331.
- Gurgul, E.; Lortz, S.; Tiedge, M.; Jorns, A.; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280.
- Plecita-Hlavata, L.; Engstova, H.; Jezek, J.; Holendova, B.; Tauber, J.; Petraskova, L.; Kren, V.; Jezek, P. Potential of Mitochondria-Targeted Antioxidants to Prevent Oxidative Stress in Pancreatic beta-cells. Oxid. Med. Cell. Longev. 2019, 2019, 1826303.
- Plecita-Hlavata, L.; Engstova, H.; Holendova, B.; Tauber, J.; Spacek, T.; Petraskova, L.; Kren, V.; Spackova, J.; Gotvaldova, K.; Jezek, J.; et al. Mitochondrial Superoxide Production Decreases on Glucose-Stimulated Insulin Secretion in Pancreatic beta Cells Due to Decreasing Mitochondrial Matrix NADH/NAD(+) Ratio. Antioxid. Redox Signal. 2020, 33, 789–815.
- Jezek, J.; Dlaskova, A.; Zelenka, J.; Jaburek, M.; Jezek, P. H(2)O(2)-Activated Mitochondrial Phospholipase iPLA(2)gamma Prevents Lipotoxic Oxidative Stress in Synergy with UCP2, Amplifies Signaling via G-Protein-Coupled Receptor GPR40, and Regulates Insulin Secretion in Pancreatic beta-Cells. Antioxid. Redox Signal. 2015, 23, 958–972.
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714.
- Appenzeller-Herzog, C.; Riemer, J.; Zito, E.; Chin, K.T.; Ron, D.; Spiess, M.; Ellgaard, L. Disulphide production by Ero1alpha-PDI relay is rapid and effectively regulated. EMBO J. 2010, 29, 3318–3329.
- Mehmeti, I.; Lortz, S.; Elsner, M.; Lenzen, S. Peroxiredoxin 4 improves insulin biosynthesis and glucose-induced insulin secretion in insulin-secreting INS-1E cells. J. Biol. Chem. 2014, 289, 26904–26913.
- Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic beta-cells. Biochim Biophys Acta Gen. Subj. 2017, 1861, 1929–1942.
- Tavender, T.J.; Sheppard, A.M.; Bulleid, N.J. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 2008, 411, 191–199.
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515.
- Hassler, J.R.; Scheuner, D.L.; Wang, S.; Han, J.; Kodali, V.K.; Li, P.; Nguyen, J.; George, J.S.; Davis, C.; Wu, S.P.; et al. The IRE1alpha/XBP1s Pathway Is Essential for the Glucose Response and Protection of beta Cells. PLoS Biol. 2015, 13, e1002277.
- Pearse, B.R.; Hebert, D.N. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 684–693.
- Plemper, R.K.; Bohmler, S.; Bordallo, J.; Sommer, T.; Wolf, D.H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 1997, 388, 891–895.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332.
- Brozzi, F.; Eizirik, D.L. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J. Med. Sci. 2016, 121, 133–139.
- Baboota, R.K.; Shinde, A.B.; Lemaire, K.; Fransen, M.; Vinckier, S.; Van Veldhoven, P.P.; Schuit, F.; Baes, M. Functional peroxisomes are required for beta-cell integrity in mice. Mol. Metab. 2019, 22, 71–83.
- Oliveira, H.R.; Verlengia, R.; Carvalho, C.R.; Britto, L.R.; Curi, R.; Carpinelli, A.R. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003, 52, 1457–1463.
- Uchizono, Y.; Takeya, R.; Iwase, M.; Sasaki, N.; Oku, M.; Imoto, H.; Iida, M.; Sumimoto, H. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci. 2006, 80, 133–139.
- Zhang, Z.; Li, J.; Yang, L.; Chen, R.; Yang, R.; Zhang, H.; Cai, D.; Chen, H. The cytotoxic role of intermittent high glucose on apoptosis and cell viability in pancreatic beta cells. J. Diabetes Res. 2014, 2014, 712781.
- Stancill, J.S.; Happ, J.T.; Broniowska, K.A.; Hogg, N.; Corbett, J.A. Peroxiredoxin 1 plays a primary role in protecting pancreatic β-cells from hydrogen peroxide and peroxynitrite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R1004–R1013.
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650.
- Wondafrash, D.Z.; Nire’a, A.T.; Tafere, G.G.; Desta, D.M.; Berhe, D.A.; Zewdie, K.A. Thioredoxin-Interacting Protein as a Novel Potential Therapeutic Target in Diabetes Mellitus and Its Underlying Complications. Diabetes Metab. Syndr. Obes. 2020, 13, 43–51.
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514.
- Nishinaka, Y.; Masutani, H.; Oka, S.; Matsuo, Y.; Yamaguchi, Y.; Nishio, K.; Ishii, Y.; Yodoi, J. Importin alpha1 (Rch1) mediates nuclear translocation of thioredoxin-binding protein-2/vitamin D(3)-up-regulated protein 1. J. Biol. Chem. 2004, 279, 37559–37565.
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146.
- Jing, G.; Westwell-Roper, C.; Chen, J.; Xu, G.; Verchere, C.B.; Shalev, A. Thioredoxin-interacting protein promotes islet amyloid polypeptide expression through miR-124a and FoxA2. J. Biol. Chem. 2014, 289, 11807–11815.
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Collins, S.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharm. 2010, 244, 77–83.
- Janjic, D.; Maechler, P.; Sekine, N.; Bartley, C.; Annen, A.S.; Wolheim, C.B. Free radical modulation of insulin release in INS-1 cells exposed to alloxan. Biochem. Pharm. 1999, 57, 639–648.
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem. 1999, 274, 27905–27913.
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791.
- Travasso, R.D.M.; Sampaio Dos Aidos, F.; Bayani, A.; Abranches, P.; Salvador, A. Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 2017, 12, 233–245.
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605.
- Kontou, M.; Will, R.D.; Adelfalk, C.; Wittig, R.; Poustka, A.; Hirsch-Kauffmann, M.; Schweiger, M. Thioredoxin, a regulator of gene expression. Oncogene 2004, 23, 2146–2152.
- Bian, M.; Fan, R.; Zhao, S.; Liu, W. Targeting the Thioredoxin System as a Strategy for Cancer Therapy. J. Med. Chem. 2019, 62, 7309–7321.
- Jastrząb, A.; Skrzydlewska, E. Thioredoxin-dependent system. Application of inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 362–371.
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Reviews. Immunol. 2020.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965.
- Schultheis, J.; Beckmann, D.; Mulac, D.; Muller, L.; Esselen, M.; Dufer, M. Nrf2 Activation Protects Mouse Beta Cells from Glucolipotoxicity by Restoring Mitochondrial Function and Physiological Redox Balance. Oxid. Med. Cell. Longev. 2019, 2019, 7518510.
- He, J.; Zhang, X.; Lian, C.; Wu, J.; Fang, Y.; Ye, X. KEAP1/NRF2 axis regulates H2O2-induced apoptosis of pancreatic beta-cells. Gene 2019, 691, 8–17.
- Cardozo, A.K.; Heimberg, H.; Heremans, Y.; Leeman, R.; Kutlu, B.; Kruhoffer, M.; Orntoft, T.; Eizirik, D.L. A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J. Biol. Chem. 2001, 276, 48879–48886.
- Meyerovich, K.; Fukaya, M.; Terra, L.F.; Ortis, F.; Eizirik, D.L.; Cardozo, A.K. The non-canonical NF-kappaB pathway is induced by cytokines in pancreatic beta cells and contributes to cell death and proinflammatory responses in vitro. Diabetologia 2016, 59, 512–521.
- Meyerovich, K.; Ortis, F.; Cardozo, A.K. The non-canonical NF-kappaB pathway and its contribution to beta-cell failure in diabetes. J. Mol. Endocrinol. 2018, 61, F1–F6.
- Lee, K.; Esselman, W.J. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic. Biol. Med. 2002, 33, 1121–1132.
- Brigelius-Flohe, R.; Flohe, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381.
- Schulze-Osthoff, K.; Beyaert, R.; Vandevoorde, V.; Haegeman, G.; Fiers, W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993, 12, 3095–3104.
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179.
- Cerf, M.E. Transcription factors regulating beta-cell function. Eur. J. Endocrinol. 2006, 155, 671–679.
- Jara, M.A.; Werneck-De-Castro, J.P.; Lubaczeuski, C.; Johnson, J.D.; Bernal-Mizrachi, E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to beta-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets 2020, 12, 32–40.
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406.
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862.
- Matsuoka, T.A.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet beta-cell function. Mol. Endocrinol. 2007, 21, 2764–2774.
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem. 2005, 280, 11107–11113.
- Kondo, T.; El Khattabi, I.; Nishimura, W.; Laybutt, D.R.; Geraldes, P.; Shah, S.; King, G.; Bonner-Weir, S.; Weir, G.; Sharma, A. p38 MAPK is a major regulator of MafA protein stability under oxidative stress. Mol. Endocrinol. 2009, 23, 1281–1290.
- El Khattabi, I.; Sharma, A. Preventing p38 MAPK-mediated MafA degradation ameliorates beta-cell dysfunction under oxidative stress. Mol. Endocrinol. 2013, 27, 1078–1090.
- Harmon, J.S.; Bogdani, M.; Parazzoli, S.D.; Mak, S.S.; Oseid, E.A.; Berghmans, M.; Leboeuf, R.C.; Robertson, R.P. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009, 150, 4855–4862.
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005, 16, 183–189.
- Dansen, T.B.; Smits, L.M.; van Triest, M.H.; de Keizer, P.L.; van Leenen, D.; Koerkamp, M.G.; Szypowska, A.; Meppelink, A.; Brenkman, A.B.; Yodoi, J.; et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009, 5, 664–672.
- De Keizer, P.L.; Burgering, B.M.; Dansen, T.B. Forkhead box o as a sensor, mediator, and regulator of redox signaling. Antioxid. Redox Signal. 2011, 14, 1093–1106.
- Burgering, B.M.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602.
- Huang, X.; Begley, M.; Morgenstern, K.A.; Gu, Y.; Rose, P.; Zhao, H.; Zhu, X. Crystal structure of an inactive Akt2 kinase domain. Structure 2003, 11, 21–30.
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233.
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H., 3rd; Wright, C.V.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Investig. 2002, 110, 1839–1847.
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163.
- Nishimura, W.; Kondo, T.; Salameh, T.; El Khattabi, I.; Dodge, R.; Bonner-Weir, S.; Sharma, A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev. Biol. 2006, 293, 526–539.
- Cao, X.; Kambe, F.; Ohmori, S.; Seo, H. Oxidoreductive modification of two cysteine residues in paired domain by Ref-1 regulates DNA-binding activity of Pax-8. Biochem. Biophys Res. Commun 2002, 297, 288–293.
- Walther, C.; Guenet, J.L.; Simon, D.; Deutsch, U.; Jostes, B.; Goulding, M.D.; Plachov, D.; Balling, R.; Gruss, P. Pax: A murine multigene family of paired box-containing genes. Genomics 1991, 11, 424–434.
- Swisa, A.; Avrahami, D.; Eden, N.; Zhang, J.; Feleke, E.; Dahan, T.; Cohen-Tayar, Y.; Stolovich-Rain, M.; Kaestner, K.H.; Glaser, B.; et al. PAX6 maintains beta cell identity by repressing genes of alternative islet cell types. J. Clin. Investig. 2017, 127, 230–243.
- Rieck, S.; Bankaitis, E.D.; Wright, C.V. Lineage determinants in early endocrine development. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2012; Volume 23, pp. 673–684.
- Bastidas-Ponce, A.; Roscioni, S.S.; Burtscher, I.; Bader, E.; Sterr, M.; Bakhti, M.; Lickert, H. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic beta-cells. Mol. Metab. 2017, 6, 524–534.
- Bensellam, M.; Jonas, J.C.; Laybutt, D.R. Mechanisms of beta-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143.
- Balakrishnan, S.; Dhavamani, S.; Prahalathan, C. beta-Cell specific transcription factors in the context of diabetes mellitus and beta-cell regeneration. Mech. Dev. 2020, 163, 103634.
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632.
- Chetboun, M.; Abitbol, G.; Rozenberg, K.; Rozenfeld, H.; Deutsch, A.; Sampson, S.R.; Rosenzweig, T. Maintenance of redox state and pancreatic beta-cell function: Role of leptin and adiponectin. J. Cell. Biochem. 2012, 113, 1966–1976.
- Ahmed Alfar, E.; Kirova, D.; Konantz, J.; Birke, S.; Mansfeld, J.; Ninov, N. Distinct Levels of Reactive Oxygen Species Coordinate Metabolic Activity with Beta-cell Mass Plasticity. Sci. Rep. 2017, 7, 3994.
- Liang, J.; Wu, S.Y.; Zhang, D.; Wang, L.; Leung, K.K.; Leung, P.S. NADPH Oxidase-Dependent Reactive Oxygen Species Stimulate beta-Cell Regeneration Through Differentiation of Endocrine Progenitors in Murine Pancreas. Antioxid. Redox Signal. 2016, 24, 419–433.
- Hoarau, E.; Chandra, V.; Rustin, P.; Scharfmann, R.; Duvillie, B. Pro-oxidant/antioxidant balance controls pancreatic beta-cell differentiation through the ERK1/2 pathway. Cell Death Dis. 2014, 5, e1487.
- Costes, S.; Broca, C.; Bertrand, G.; Lajoix, A.D.; Bataille, D.; Bockaert, J.; Dalle, S. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: A key role in glucose-mediated pancreatic beta-cell survival. Diabetes 2006, 55, 2220–2230.
- Hussain, M.A.; Porras, D.L.; Rowe, M.H.; West, J.R.; Song, W.J.; Schreiber, W.E.; Wondisford, F.E. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Mol. Cell. Biol. 2006, 26, 7747–7759.
- Piera-Velazquez, S.; Hawkins, D.F.; Whitecavage, M.K.; Colter, D.C.; Stokes, D.G.; Jimenez, S.A. Regulation of the human SOX9 promoter by Sp1 and CREB. Exp. Cell Res. 2007, 313, 1069–1079.
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71.
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat. Cell Biol. 2006, 8, 501–508.
- Fruhbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20.
- Lee, Y.H.; Magkos, F.; Mantzoros, C.S.; Kang, E.S. Effects of leptin and adiponectin on pancreatic beta-cell function. Metabolism 2011, 60, 1664–1672.
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Hurley, J.D.; Smith, D.M.; Ghatei, M.A.; Withers, D.J.; Gardiner, J.V.; Bailey, C.J.; Bloom, S.R. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Investig. 1997, 100, 2729–2736.
- Kieffer, T.J.; Heller, R.S.; Leech, C.A.; Holz, G.G.; Habener, J.F. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 1997, 46, 1087–1093.
- Kuehnen, P.; Laubner, K.; Raile, K.; Schofl, C.; Jakob, F.; Pilz, I.; Path, G.; Seufert, J. Protein phosphatase 1 (PP-1)-dependent inhibition of insulin secretion by leptin in INS-1 pancreatic beta-cells and human pancreatic islets. Endocrinology 2011, 152, 1800–1808.
- Sim, A.T.; Baldwin, M.L.; Rostas, J.A.; Holst, J.; Ludowyke, R.I. The role of serine/threonine protein phosphatases in exocytosis. Biochem. J. 2003, 373, 641–659.
- Staiger, K.; Stefan, N.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Machicao, F.; Kellerer, M.; Stumvoll, M.; Fritsche, A.; et al. Adiponectin is functionally active in human islets but does not affect insulin secretory function or beta-cell lipoapoptosis. J. Clin. Endocrinol. Metab. 2005, 90, 6707–6713.
- Llanos, P.; Contreras-Ferrat, A.; Barrientos, G.; Valencia, M.; Mears, D.; Hidalgo, C. Glucose-Dependent Insulin Secretion in Pancreatic beta-Cell Islets from Male Rats Requires Ca2+ Release via ROS-Stimulated Ryanodine Receptors. PLoS ONE 2015, 10, e0129238.
- Jansson, D.; Ng, A.C.; Fu, A.; Depatie, C.; Al Azzabi, M.; Screaton, R.A. Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc. Natl. Acad. Sci. USA 2008, 105, 10161–10166.
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocana, A. Human beta-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831.
- Sato, Y.; Endo, H.; Okuyama, H.; Takeda, T.; Iwahashi, H.; Imagawa, A.; Yamagata, K.; Shimomura, I.; Inoue, M. Cellular hypoxia of pancreatic beta-cells due to high levels of oxygen consumption for insulin secretion in vitro. J. Biol. Chem. 2011, 286, 12524–12532.
- Zhdanov, A.V.; Ward, M.W.; Prehn, J.H.; Papkovsky, D.B. Dynamics of intracellular oxygen in PC12 Cells upon stimulation of neurotransmission. J. Biol. Chem. 2008, 283, 5650–5661.
- O’Hagan, K.A.; Cocchiglia, S.; Zhdanov, A.V.; Tambuwala, M.M.; Cummins, E.P.; Monfared, M.; Agbor, T.A.; Garvey, J.F.; Papkovsky, D.B.; Taylor, C.T.; et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2188–2193.
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518.
- Olsson, R.; Carlsson, P.O. A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes 2011, 60, 2068–2075.
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448.
- Yasui, S.; Mawatari, K.; Morizumi, R.; Furukawa, H.; Shimohata, T.; Harada, N.; Takahashi, A.; Nakaya, Y. Hydrogen peroxide inhibits insulin-induced ATP-sensitive potassium channel activation independent of insulin signaling pathway in cultured vascular smooth muscle cells. J. Med. Investig. 2012, 59, 36–44.
- Sakaguchi, R.; Mori, Y. Transient receptor potential (TRP) channels: Biosensors for redox environmental stimuli and cellular status. Free Radic. Biol. Med. 2020, 146, 36–44.
- Finol-Urdaneta, R.K.; Remedi, M.S.; Raasch, W.; Becker, S.; Clark, R.B.; Struver, N.; Pavlov, E.; Nichols, C.G.; French, R.J.; Terlau, H. Block of Kv1.7 potassium currents increases glucose-stimulated insulin secretion. EMBO Mol. Med. 2012, 4, 424–434.
- MacDonald, P.E.; Salapatek, A.M.; Wheeler, M.B. Temperature and redox state dependence of native Kv2.1 currents in rat pancreatic beta-cells. J. Physiol. 2003, 546, 647–653.
- Mittal, M.; Gu, X.Q.; Pak, O.; Pamenter, M.E.; Haag, D.; Fuchs, D.B.; Schermuly, R.T.; Ghofrani, H.A.; Brandes, R.P.; Seeger, W.; et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2012, 52, 1033–1042.
- Gerst, J.E. SNARE regulators: Matchmakers and matchbreakers. Biochim Biophys Acta 2003, 1641, 99–110.
- Ivarsson, R.; Quintens, R.; Dejonghe, S.; Tsukamoto, K.; Renstrom, E.; Schuit, F.C. Redox control of exocytosis: Regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 2005, 54, 2132–2142.
- Reinbothe, T.M.; Ivarsson, R.; Li, D.Q.; Niazi, O.; Jing, X.; Zhang, E.; Stenson, L.; Bryborn, U.; Renstrom, E. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol. Endocrinol. 2009, 23, 893–900.
- Ferdaoussi, M.; Dai, X.; Jensen, M.V.; Wang, R.; Peterson, B.S.; Huang, C.; Ilkayeva, O.; Smith, N.; Miller, N.; Hajmrle, C.; et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional beta cells. J. Clin. Investig. 2015, 125, 3847–3860.
- Xu, Z.; Lam, L.S.; Lam, L.H.; Chau, S.F.; Ng, T.B.; Au, S.W. Molecular basis of the redox regulation of SUMO proteases: A protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008, 22, 127–137.
- Ferdaoussi, M.; MacDonald, P.E. Toward Connecting Metabolism to the Exocytotic Site. Trends Cell Biol. 2017, 27, 163–171.
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.M. Thiol switches in membrane proteins - Extracellular redox regulation in cell biology. Biol. Chem. 2020.
- Lundberg, M.; Fernandes, A.P.; Kumar, S.; Holmgren, A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem. Biophys. Res. Commun. 2004, 319, 801–809.
- Xiong, B.; Jha, V.; Min, J.K.; Cho, J. Protein disulfide isomerase in cardiovascular disease. Exp. Mol. Med. 2020, 52, 390–399.
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion. Mol. Med. 2015, 21, 98–108.
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of β-cell function by thioredoxin. Redox Biol. 2020, 34, 101570.
- Jikimoto, T.; Nishikubo, Y.; Koshiba, M.; Kanagawa, S.; Morinobu, S.; Morinobu, A.; Saura, R.; Mizuno, K.; Kondo, S.; Toyokuni, S.; et al. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol. Immunol. 2002, 38, 765–772.
- Kakisaka, Y.; Nakashima, T.; Sumida, Y.; Yoh, T.; Nakamura, H.; Yodoi, J.; Senmaru, H. Elevation of serum thioredoxin levels in patients with type 2 diabetes. Horm. Metab. Res. 2002, 34, 160–164.
- Asami, K.; Inagaki, A.; Imura, T.; Sekiguchi, S.; Fujimori, K.; Masutani, H.; Yodoi, J.; Satomi, S.; Ohuchi, N.; Goto, M. Thioredoxin-1 attenuates early graft loss after intraportal islet transplantation in mice. PLoS ONE 2013, 8, e70259.
- Willems, S.H.; Tape, C.J.; Stanley, P.L.; Taylor, N.A.; Mills, I.G.; Neal, D.E.; McCafferty, J.; Murphy, G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem. J. 2010, 428, 439–450.
- Bass, R.; Edwards, D.R. ADAMs and protein disulfide isomerase: The key to regulated cell-surface protein ectodomain shedding? Biochem. J. 2010, 428, e3–e5.
- Düsterhöft, S.; Jung, S.; Hung, C.W.; Tholey, A.; Sönnichsen, F.D.; Grötzinger, J.; Lorenzen, I. Membrane-proximal domain of a disintegrin and metalloprotease-17 represents the putative molecular switch of its shedding activity operated by protein-disulfide isomerase. J. Am. Chem. Soc. 2013, 135, 5776–5781.
- Pedersen, K.B.; Chodavarapu, H.; Porretta, C.; Robinson, L.K.; Lazartigues, E. Dynamics of ADAM17-Mediated Shedding of ACE2 Applied to Pancreatic Islets of Male db/db Mice. Endocrinology 2015, 156, 4411–4425.
- Chhabra, K.H.; Chodavarapu, H.; Lazartigues, E. Angiotensin converting enzyme 2: A new important player in the regulation of glycemia. IUBMB Life 2013, 65, 731–738.
- Bergerhausen, L.; Grosche, J.; Meißner, J.; Hecker, C.; Caliandro, M.F.; Westerhausen, C.; Kamenac, A.; Rezaei, M.; Mörgelin, M.; Poschmann, G.; et al. Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants 2020, 9, 227.
- Passam, F.; Chiu, J.; Ju, L.; Pijning, A.; Jahan, Z.; Mor-Cohen, R.; Yeheskel, A.; Kolšek, K.; Thärichen, L.; Aponte-Santamaría, C.; et al. Mechano-redox control of integrin de-adhesion. eLife 2018, 7.
- Townsend, S.E.; Gannon, M. Extracellular Matrix-Associated Factors Play Critical Roles in Regulating Pancreatic β-Cell Proliferation and Survival. Endocrinology 2019, 160, 1885–1894.
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687.
- Alam, N.; Goel, H.L.; Zarif, M.J.; Butterfield, J.E.; Perkins, H.M.; Sansoucy, B.G.; Sawyer, T.K.; Languino, L.R. The integrin-growth factor receptor duet. J. Cell. Physiol. 2007, 213, 649–653.
- Xu, S.Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC channel activation by extracellular thioredoxin. Nature 2008, 451, 69–72.
- Islam, M.S. Molecular Regulations and Functions of the Transient Receptor Potential Channels of the Islets of Langerhans and Insulinoma Cells. Cells 2020, 9, 685.

