 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudia Ricci | + 3959 word(s) | 3959 | 2021-04-20 09:46:13 | | | |
| 2 | Camila Xu | Meta information modification | 3959 | 2021-04-21 10:40:03 | | |
Video Upload Options
Cerebral cavernous malformations (CCMs): an overall view from genes to endothelial cells.
1. Introduction
Cerebral cavernous malformation (CCM) is a disease predominantly affecting neurovasculature. These malformations consist of densely packed, enlarged capillary “caverns” generally arranged in a back-to-back pattern with no involvement of brain parenchyma [1]. Histologically, the lesions lack smooth muscle, elastic tissue, and all of the elements required for mature blood vessels. Clustered capillaries are lined by a single layer of endothelium included in a dense collagen matrix, and so characterized by an inhomogeneous vessel wall because of the presence of defective cell–cell junctions. This condition results in the impairment of the blood–brain barrier that predisposes it to episodes of thrombosis and bleeding. Due to the cerebral hemorrhages, patients may present an increased risk for stroke, seizures, motor and sensory deficits, and non-specific headaches [2][3]. However, many affected individuals are clinically asymptomatic during their entire life [4]. Apart from the brain and spinal cord location, CCMs are also seen in the retina, liver, kidney, and as hyperkeratotic cutaneous capillaryvenous malformations on the skin [3][5].
The penetrance has been calculated to be 0.5% in the general population worldwide (1 in 200–250 individuals), but the clinical prevalence is much lower because up to 50% of individuals with CCM are asymptomatic [6][7]. CCM-related clinical manifestations usually appear between the third and fifth decades of life, with no gender predominance, even if some cases have been also reported in children [8]. Clinical signs and symptoms are largely determined by the location, number, and size (from a few millimeters to several centimeters) of the lesions. Given that both size and the number of malformations can change over time, new signs and symptoms can arise at every stage of a patient’s life. The 3T Magnetic Resonance Imaging represents the gold standard for CCM detection [9]. Treatment differs between the administration of antiepileptic drugs in patients with seizures and surgical CCM resection in symptomatic patients, keeping in mind that lesions are not always easily accessible [6][10].
2. CCM Pathogenesis
Two forms of the disorder have been identified—sporadic (80%) and inherited (20%) disease. Sporadic cases, which occur in people with no family history of CCM, are mostly associated with the presence of a single lesion, while multiple lesions are frequently detected in familial CCM (fCCM) [11].
To date, both fCCM and sporadic forms have been attributed to mutations in three different genes, identified by linkage analysis and known as CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10.
The fCCM form has an autosomal dominant inheritance, with incomplete penetrance and variable expressivity [12]. The Knudsonian two-hit mechanism is the most accredited hypothesis to explain CCM pathogenesis [13]. According to this theory, loss of one allele due to a germline mutation in all cells (first hit) is followed by the occurrence, just in some cells, of a somatic mutation in the other allele (second hit), triggering the initiation of the lesions [11][14]. Animal experiments demonstrate that knockout mice for CCM genes die during embryogenesis because of heart deficiency and vascular defects, while mice carrying only one mutated allele rarely develop cavernous malformations [15]. These findings suggest that the loss of both copies of the gene represents the driving force for CCM pathogenesis.
Nevertheless, the definitive cause that determines CCMs remains an open question. In addition to this evidence, another important feature of the CCM condition is the heterogeneity of the phenotypes. More than 350 distinct CCM1/CCM2/CCM3 mutations have been published to date [16], and patients carrying different mutations in different genes are clinically and phenotypically indistinguishable, although CCM3 patients are often more severely affected with earlier symptomatic onset [17].
Of note, almost 5–15% of fCCMs do not show mutations in the three known CCM genes, raising the possibility of the existence of an additional gene correlated to the disease [18][19]. However, more than 15 years after the identification of the third CCM gene [18], this hypothesis is rather unlikely. It is more probable that the CCM families apparently negative for mutations in the three known CCM genes harbor a pathogenic variant not identified by the routinely used techniques [9].
All of the three discovered genes associated with CCMs found in mammals, vertebrates, and simple organisms (i.e., Caenorhabditis elegans), are well conserved among species, giving the advantage of exploiting numerous animal models to progress in the knowledge of this pathological condition [20].
2.1. CCM1 Gene
CCM1/KRIT1 (OMIM#604214) was the first gene implicated in CCM to be identified on chromosome 7 locus q21.2. The coding region is represented by 16 of the 19 total exons that constitute the gene [21]. KRIT1 is ubiquitously expressed in embryos with a pronounced rate in large vessels, while it progressively scales down in adulthood, becoming detectable predominantly in the nervous and epithelial tissues [22]. Knockout mice for this gene die at midgestation because of cardiac defects and vascular anomalies [23], showing the importance of KRIT1 in vessel development and morphogenesis.
Mutations in this gene have been found in nearly all Hispanic American patients [24] and globally account for up to 50% of familial cases [25]. The clinical penetrance is estimated to be about 88% [1] and over 300 distinct mutations have been identified in this gene until now [16]. Frameshift, nonsense, missense, and splice site sequence variants are the most common mutations occurring in KRIT1, all resulting in early stop codons and, subsequently, in truncated and improperly working proteins. Moreover, rare insertions or deletions have also been identified [26][27][28][29][30][31].
2.2. CCM2 Gene
The second CCM locus, also known as MGC4607 or C7orf22 (OMIM#607929), identified on 7p13, consists of 10 coding exons [32]. Almost 20% of familial CCMs are due to mutations in this gene that was reported to be the only one with 100% of clinical and radiological penetrance [33]. However, recently, for the first time, a penetrance <100%, equivalent to 70%, has been reported for the CCM2 gene [3]. More than 90 distinct mutations (missense, nonsense, frameshift, and splice site variants) have been found in the CCM2 gene up till now [16].
As KRIT1, CCM2 is ubiquitously expressed in the endothelium of various organs [34] and CCM2-silencing in zebrafish [35] and mice [36] revealed defects in arteries and veins formation, determining death during embryonal stages. Interestingly, mice heterozygous for KRIT1 or CCM2 do not develop lesions, unless the occurrence of a secondary hit, as reported by Shenkar et al. [37].
A lower number of lesions are commonly detected in CCM2-mutated patients in comparison to carriers of mutations in the other CCM genes, and the risk of an increase of the number of lesions during life is lower than in patients with mutations in the KRIT1 gene [19]. In addition, hyperkeratotic cutaneous capillaryvenous malformations have been reported to be strongly associated with KRIT1 variants, while patients with mutations in the CCM2 gene are less prone to develop cutaneous lesions [38].
2.3. CCM3 Gene
CCM3, also named PDCD10 (programmed cell death 10) (OMIM#609118), is the most recently discovered gene associated with CCM, identified by Bergametti et al. in 2005 after screening of patients with no mutations in KRIT1 or CCM2 [18]. PDCD10 is a highly conserved pro-apoptotic gene, located on 3q26.1, containing seven coding and three non-coding exons. As reported for the other two genes associated with CCM disease, all the over 70 mutations identified in PDCD10 result in protein loss of function [16]. Patients carrying mutations in the PDCD10 gene are less common than KRIT1 or CCM2 carriers; only 10% of all familial CCM cases are due to mutations in this gene, with a penetrance of about 63% [39]. However, the clinical phenotype of patients with PDCD10 mutations is often significantly more severe and symptoms onset occurs earlier in life [17]. PDCD10 deletion in animal model causes aberrant vasculogenesis and hematopoiesis [40].
2.4. Any Other Additional Genes?
As previously reported, patients with no mutations in KRIT1, CCM2, and PDCD10 genes might carry pathogenic variants in different genetic loci. Thus, the existence of an additional gene associated with CCM has been postulated. In addition to the three known genes, in 2008, Gianfrancesco et al. [41] proposed the zona pellucida-like domain containing 1 (ZPLD1) gene (3q12.3) as a new possible candidate implicated in CCM disease. However, further studies described this as a rare condition because the ZPLD1 gene does not directly cause CCM, but may be implicated in some unidentified regulatory pathway associated with the disease [21][42]. Genetic screening currently includes the analysis of coding exons and exon/intron junctions followed by sequencing and deletion/duplication testing of KRIT1, CCM2, and PDCD10. Investigation on new genes responsible for CCM pathology is still ongoing, but it seems to be more likely that the familial cases negative for mutations in the three genes actually carry a pathogenic variant not identified by the commonly used diagnostic methods (e.g., a variant outside the screened exonic regions, deep intron variants, or a copy number neutral genomic rearrangement in one of the three genes) [9].
Key information of the three CCM genes is summarized in Table 1.
Table 1. Summary of cerebral cavernous malformation (CCM) genes and relative proteins. The number of identified mutations is referred to data included in the Human Gene Mutation Database (HGMD) [31].
| Gene. | OMIM | Genetic Locus |
Protein | UniProt | Identified Mutations |
|---|---|---|---|---|---|
| CCM1/KRIT1 | #604214 | 7q21.2 | KRIT1 | #O00522 | >300 |
| CCM2/MGC4607 | #607929 | 7p13 | Malcavernin | #Q9BSQ5 | >90 |
| CCM3/PDCD10 | #609118 | 3q26.1 | PDCD10/TFAR15 | #Q9BUL8 | >70 |
3. CCM Proteins
The inherited nature of CCM and the discovery of disease-related genes sparked interest in investigating their functional roles. The ubiquitous expression of the three CCM genes in various cells and tissues provides evidence for their contribution in several physiological and pathological conditions [43]. However, the cellular type most strongly associated with cavernous malformations is the endothelial cell. The role played by CCM genes in other cellular models strictly associated with lesions localization (i.e., neuronal cells, astrocytes, pericytes, and smooth muscle cells) is unknown, and there is no evidence about their direct contributions to CCM pathology [44]. There is probably a tight interplay between endothelial cells and the glia in the central nervous system (CNS), which may explain why mutations in CCM-related genes are followed by the appearance of lesions predominantly localized in the brain and spinal cord [45].
KRIT1, CCM2, and PDCD10 genes encode for proteins different in structure and not sharing sequence homology. Each gene product is a multi-domain adaptor protein, which interacts with numerous binding molecules and participates in several different signaling pathways. On the other hand, KRIT1, CCM2, and PDCD10 proteins can exist into a heterotrimeric complex, the CCM signaling complex (CSC), being partners in numerous cellular processes that significantly affect CCM development and pathogenesis [46]. Thus, CCM proteins fulfill critical roles in many cellular events, such as cell polarity, cytoskeletal reorganization, cell proliferation, cellular adhesion, and migration, impacting angiogenesis, cell–cell junction integrity, vascular permeability, and apoptosis, whether as part of the ternary complex or not.
In the present review, each CCM protein and its related functions are described in a specific paragraph, while the converging molecular pathways and the role of the trimeric CCM complex are depicted in the last section.
3.1. KRIT1 Protein
KRIT1 gene encodes for the largest (84 kDa) and best-characterized of the three CCM proteins; named Krev interaction trapped protein 1 (UniProt #O00522), it is a 736-amino acid sequence without catalytic activity that can be found in many different subcellular sites. Several insights about KRIT1 functions have been obtained carrying out an extensive analysis of the structural motifs contained in the protein. KRIT1 consists of an N-terminal NUDIX domain, followed by three NPxY/F (Asn-Pro-X-Tyr/Phe) motifs, a central ankyrin repeat region (ARD), and a C-terminal FERM domain (band 4.1 ezrin radixin moesin domain) [47][48] (Figure 1).
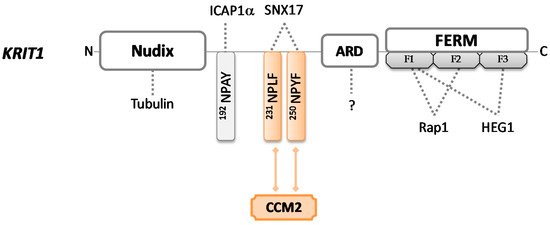
Figure 1. Schematic representation of KRIT1 protein. KRIT1 consists of a Nudix domain, three NPxY/F sites, four ankyrin repeat domains, and a FERM domain. FERM contains three subdomains (F1, F2, and F3) and each of them folds differently—F1 adopts a ubiquitin-like fold, F2 adopts an acyl–CoA binding protein fold, and F3 adopts a PTB domain-like fold. Colored forms and lines in the figure indicate domains and interactions required to bind CCM2 in order to build the CCM trimeric complex. Dotted lines indicate intermolecular interactions that may occur through each protein domain.
The FERM module consists of three subdomains (F1, F2, and F3). In 1997, Serebriiskii et al. [49] firstly identified KRIT1 as a binding partner for Rap1, a member of the RAS family of GTPases, which plays important roles in both intercellular and cell–extra-cellular matrix (ECM) adhesion, as well as in cell proliferation pathways. Later, in 2012, it was demonstrated that the binding occurs via KRIT1 FERM domain [50] through F1 and F2 subdomains, while the F1–F3 interface forms a hydrophobic pocket that may interact with the cytoplasmic tail of the transmembrane heart of glass (HEG1) protein [51]. HEG1 is critical for the proper localization of KRIT1 at endothelial cell–cell junctions, contributing to junctional stability and participating in signaling due to the Rap1–KRIT1 complex [52]. KRIT1 mutants that cannot recruit Rap1 or HEG1 are associated with cardiovascular defects [51], suggesting that the set of these interactions plays an important role in the stabilization of cell–cell junctions and may account for KRIT1 involvement in CCM pathogenesis [53].
The three NPxY/F motifs mediate the interactions with other binding partners. The unique NPxY module interacts with the phosphotyrosine-binding (PTB) domain of a protein known as α-isoform of integrin cytoplasmic-associated protein-1 (ICAP1α), which mainly works as a suppressor of β1 integrin. ICAP1α competes with the integrin activators talin and kindlin, in order to modulate integrin activity and focal adhesion turnover. If talin and kindlin recruit actin bundles and favor cell–ECM attachment, ICAP1α inhibits the integrin–actin axis, disrupting cell adhesion and migration [54]. KRIT1 acts as a competitive inhibitor for the interaction between ICAP1α and β1 integrin. Thus, because ICAP1α utilizes the same PTB site to bind KRIT1 and the cytoplasmic portion of β1 integrin when it is bound to KRIT1, it cannot interact with, and consequently, negatively regulate the integrin activity, resulting in increased integrin-mediated cell adhesion and hyperangiogenesis [34][54]. Furthermore, ICAP1α exhibits a stabilizing function toward KRIT1 and induces its translocation into the nucleus, preventing proteasomal degradation. This evidence highlights how KRIT1 can fulfill such a dual role (cytoplasmic and nuclear), but its exact function in the nucleus remains at present completely unknown [55]. Of note, KRIT1 may provide potential crosstalk between integrin and Rap1 signaling by communicating in concert with ICAP1α and Rap1, also suggesting the involvement of the latter protein in integrin-mediated adhesive events. Importantly, mutations in the KRIT1 gene may nullify this molecular association, confirming the crucial implication of KRIT1 in cell–cell and cell-matrix adhesion [56][57].
Through the NPxF motifs, KRIT1 recruits different binding proteins, including CCM2. KRIT1 and CCM2 show similar spatiotemporal expression patterns, demonstrating that they are strictly interconnected [58]. It is worth noting that CCM2 binding to KRIT1 does not abrogate KRIT1–ICAP1α interaction, supporting the hypothesis that a ternary ICAP1α/KRIT1/CCM2 complex may exist. In 2005, Zawistowski et al. [59] reported that the subcellular localization of KRIT1 is differentially influenced by these two proteins—if ICAP1α induces nuclear translocation, CCM2 maintains KRIT1 into the cytosol. However, it has been recently suggested that the whole complex ICAP1α/KRIT1/CCM2 shuttles back and forth from cytoplasm to nucleus and plays different roles in different cellular compartments [44].
KRIT1 uses NPxF motifs also to recruit sorting nexin 17 (SNX17) and localize into intracellular vesicles, but the effects produced by this interaction remain to be elucidated [60].
Furthermore, the NPxY/F region supports changes in protein shape, which in turn define the functional outputs. In fact, KRIT1 functions depend on the conformation that the protein acquires in subcellular locations—an open conformation enables the KRIT1-ICAPα interaction [61], while a closed conformation mainly exposes the NUDIX region [62].
The N-terminal NUDIX domain, recently described as pseudo-NUDIX because it is lacking hydrolase activity normally showed by NUDIX domains, is responsible for the binding of KRIT1 to tubulin, making KRIT1 a microtubule-associated protein. Therefore, KRIT1 modulates the cytoskeletal organization of endothelial cells, playing a crucial role also in cell morphology. Gunel et al. [10] pointed out that the loss of KRIT1 results in impaired tubulogenesis and consequently leads to abnormal vessel development, which is typical of the CCM lesions.
Finally, four ankyrin repeat domains are functionally available for the interaction with several molecules of different origins and natures [63]. Their precise roles in CCM pathogenesis remain elusive, but interestingly, the combination of ankyrin repeats with the FERM domain is apparently unique to KRIT1 [57], suggesting that it is probable that further functions of this protein have yet to be discovered.
Collectively, KRIT1 regulates several cellular processes and plays numerous pivotal roles in different signaling pathways when assembled with CCM2 and PDCD10 into the CSC.
3.2. CCM2 Protein
The CCM2 gene encodes for a 444-amino acid product referred to as CCM2 or malcavernin (48 kDa) (UniProt #Q9BSQ5), a scaffolding protein without any enzymatic activity. Structurally, from N-terminus to C-terminus, malcavernin consists of a PTB domain, an LD-like motif, and a helical harmonin homology domain (HHD) (Figure 2).
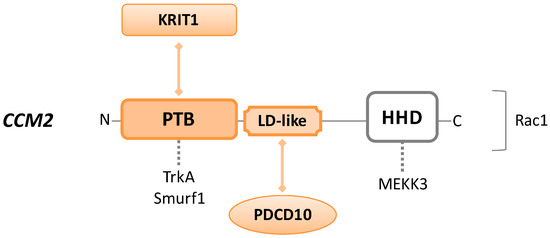
Figure 2. Schematic representation of CCM2/malcavernin protein. CCM2 consists of an N-terminal phosphotyrosine-binding (PTB) domain, a C-terminal harmonin homology domain (HHD), and a central LD-like motif. Colored forms and lines in the figure indicate domains and interactions required to build the CCM trimeric complex. Dotted lines indicate intermolecular interactions that may occur through each protein domain. Rac1 is placed on the right of the CCM2 sequence because the precise binding site is unknown.
Malcavernin is central to the CCM trimeric complex organization, as the PTB and LD-like motif interact with NPxF of KRIT1 and focal adhesion targeting homology (FAT-H) domain of PDCD10, respectively. In this manner, CCM2 works as a linker, bringing together KRIT1 and PDCD10, which otherwise have no affinity for each other [2]. Loss of function mutations in any of these binding domains disrupt the CSC, alter the integrity at cell–cell junctions, and cause aberrant cell–ECM adhesion [64].
However, KRIT1 and CCM2 are more strictly connected to each other than to PDCD10. As mentioned before, KRIT1–CCM2 interaction is essential for CCM2 localization at the cell–cell junctions, and for the maintenance of KRIT1 into the cytoplasm. Interestingly, CCM2 depletion leads to the loss of KRIT1 from the cell membrane, inducing endothelial barrier dysfunction [65]. Moreover, several studies describe the crucial involvement of CCM2 in the KRIT1–HEG1 pathway, required for normal cardiovascular development [53][66][67].
The CCM2–PTB domain is also able to bind Tropomyosin receptor kinase A (TrkA), known to be involved in apoptosis, and Smad ubiquitin regulatory factor 1 (Smurf1), an upstream RhoA signaling protein [68].
Among other functions, CCM2 can recruit Rac1 protein, interfering with the actin cytoskeleton, or bind to MEKK3 (mitogen-activated protein kinase (MAPK) kinase kinase 3) via the HHD region, affecting its signaling cascade. MEKK3 changes cell morphology in response to the hyperosmotic shock and regulates endothelial to mesenchymal transition (EndMT), a condition that shows characteristics frequently seen in CCM lesions (loss of integrity in cell–cell junctions and increased cell migration and proliferation activity) [69].
The aforementioned interactions of CCM2 with Smurf1, Rac1, and MEKK3 affect signaling cascades that are also influenced by KRIT1 or PDCD10, but it is still not well characterized whether CCM proteins interfere with the pathways as part of the CSC or not. These converging molecular activities will be deeply explained later.
Recently, Zheng et al. [70] identified a paralog of malcavernin, termed CCM2 protein-like (CCM2L), the sequence of which is highly similar, but not entirely homologous, to CCM2 and which displays some features different from CCM2. First of all, the expression of CCM2L is restricted to endothelial cells, in contrast to CCM2, which is ubiquitously expressed. As CCM2L lacks an LD-like motif for binding to PDCD10, it directly competes with CCM2 for binding to KRIT1 but cannot work as the hub of CSC, and consequently inhibits the activities mediated by the KRIT1/CCM2/PDCD10 trimeric complex. However, in some cases, CCM2L can compensate for many functions of malcavernin. For example, it can inhibit the nuclear translocation of KRIT1, maintaining the protein into the cytosol, induce cell death by activating TrkA via the C-term PTB domain, and work as an osmosensing scaffold protein (OSM) for the MEKK3 pathway [71]. Even though the exact functions of CCM2L are still poorly defined, it is clear that this CCM2 homolog has some roles complementary to malcavernin in CCM pathogenesis.
3.3. CCM3 Protein
The PDCD10 gene encodes for PDCD10, a protein also named TF-1 cell apoptosis-related protein (TFAR15) (UniProt #Q9BUL8). It is a 25kDa protein consisting of 212 amino acids and composed of two main domains: An N-terminal dimerization domain and a focal adhesion targeting homology (FAT–H) domain, localized at the C-terminus of the protein [72] (Figure 3).
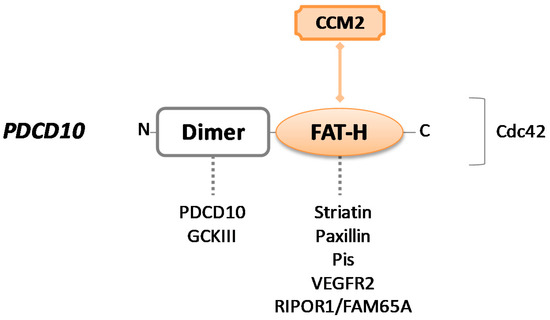
Figure 3. Schematic representation of CCM3/PDCD10 protein. PDCD10 consists of a dimer site and a focal adhesion targeting homology (FAT–H) domain. Colored forms and lines in the figure indicate the crucial interaction with CCM2 to build the CCM trimeric complex. Dotted lines indicate intermolecular interactions that may occur through each protein domain. The binding site for the Cdc42 protein is unknown.
The hydrophobic patch 1 (HP1) region of α-helical FAT–H domain is responsible for the crucial interaction between PDCD10 and the LD motif of CCM2 for the assembly of CSC [73]. Although the interaction between CCM2 and PDCD10 has stabilizing effects for all the proteins of the complex, protecting them from degradation, PDCD10 predominantly operates outside of the CCM signaling complex.
The best-known function singularly mediated by PDCD10 involves its dimerization domain, which consists of four α-helices and alternatively supports homodimerization (with PDCD10 itself) and heterodimerization with the striatin interacting phosphatase and kinase (STRIPAK) complex. STRIPAK components are represented by striatins and striatin-interacting proteins STRIP1/2, protein phosphatase 2A (PP2A), protein kinases belonging to the germinal center kinase III (GCKIII) family (i.e., STK25, STK24, and MST4), etc. [74][75][76].
The STRIPAK complex is a central regulator node of several processes in endothelial cells. First of all, it is crucial for Golgi assembly and positioning during cell migration, and PDCD10 has been shown to actively participate in this process, stabilizing the GCKIII kinases through anchorage to GM130 cisGolgi-resident protein and promoting proper Golgi orientation [77]. Of note, the loss of PDCD10 downregulates STK25 activity, leading to the aberrant repositioning of both Golgi apparatus and centrosome toward the leading edge of the cell, consequently disrupting cellular directional migration [78]. Then, as a STRIPAK component, PDCD10 plays a crucial role as a dual regulator of exocytosis. It can recruit STK24, which is known as an exocytosis inhibitor, neutralizing it to enhance exocytosis, while PDCD10-UNC13 interaction has the opposite effect [79]. This regulatory function of PDCD10 is essential in endothelial cells where excessive secretion of angiopoietin-2 (ANGPT-2) to the extracellular space strongly impacts junctional and vascular stability [80][81]. Lastly, the MST4 kinase of the GCKIII family interacts with PDCD10 under conditions of oxidative stress to regulate cell polarity and the NF–kB signaling pathway [82].
Using the FAT–H domain, PDCD10 may form other dynamic interactions apart from CCM trimeric complex. It can recruit striatins and paxillin or bind to phosphotidylinositides (PIs) that facilitate its translocation from the Golgi to the plasma membrane [83]. Through the same binding region, PDCD10 contributes to angiogenesis and cell proliferation interacting with VEGFR2—it has been described that PDCD10 mutants show a reduced VEGFR2 expression because of pronounced endocytosis, resulting in aberrant vascular development and cell death [40]. Recently, it was reported that PDCD10–FAT–H can also directly bind to Rho family interacting cell polarization regulator 1 (RIPOR1), participating in cellular morphology and migration via RhoA signaling [84].
PDCD10, as its name suggests, possesses pro-apoptotic activity. As already described, CCM2 is involved in apoptosis through the binding to TrkA and, most of the time, this interaction happens simultaneously to PDCD10–STK25 association, forming a large complex involved in apoptosis [85]. However, PDCD10 can lead to apoptosis through an alternative mechanism, by activation of the caspase-3 pathway [86]. In addition, pro-survival effects have also been reported for this protein [87].
Moreover, PDCD10 importantly contributes to cellular proliferation. Interestingly, overexpression of PDCD10 in CCM2-deficient cells restore the cells’ ability to proliferate, while upregulation of CCM2 in PDCD10-defective cells does not rescue proliferative activity [73]. PDCD10 loss also results in defective cell migration, due to suppression of RhoA signaling that dysregulates cytoskeleton organization [88].
References
- Revencu, N.; Vikkula, M. Cerebral cavernous malformation: New molecular and clinical insights. J. Med. Genet. 2006, 43, 716–721.
- Faurobert, E.; Albiges-Rizo, C. Recent insights into cerebral cavernous malformations: A complex jigsaw puzzle under construction. FEBS J. 2010, 277, 1084–1096.
- Scimone, C.; Donato, L.; Katsarou, Z.; Bostantjopoulou, S.; D’Angelo, R.; Sidoti, A. Two novel KRIT1 and CCM2 mutations in patients affected by cerebral cavernous malformations: New information on CCM2 penetrance. Front. Neurol. 2018, 9, 953.
- Sahoo, T.; Johnson, E.W.; Thomas, J.W.; Kuehl, P.M.; Jones, T.L.; Dokken, C.G.; Touchman, J.W.; Gallione, C.J.; Lee-Lin, S.Q.; Kosofsky, B.; et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 1999, 8, 2325–2333.
- Feldmeyer, L.; Baumann-Vogel, H.; Tournier-Lasserve, E.; Riant, F.; Jung, H.H.; French, L.E.; Kamarashev, J. Hyperkeratotic cutaneous vascular malformation associated with familial cerebral cavernous malformations (FCCM) with KRIT1/CCM1 mutation. Eur. J. Dermatol. 2014, 24, 255–257.
- Krisht, K.M.; Whitehead, K.J.; Niazi, T.; Couldwell, W.T. The pathogenetic features of cerebral cavernous malformations: A comprehensive review with therapeutic implications. Neurosurg. Focus 2010, 29, 2.
- Akers, A.; Al-Shahi Salman, R.; Awad, I.A.; Dahlem, K.; Flemming, K.; Hart, B.; Kim, H.; Jusue-Torres, I.; Kondziolka, D.; Lee, C.; et al. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: Consensus recommendations based on systematic literature review by the angioma alliance scientific advisory board clinical experts panel. Neurosurgery 2017, 80, 665–680.
- Morrison, L.; Akers, A. Cerebral Cavernous Malformation, Familial; GeneReviews [Internet]: Seattle, WA, USA, 2016.
- Spiegler, S.; Rath, M.; Paperlein, C.; Felbor, U. Cerebral cavernous malformations: An update on prevalence, molecular genetic analyses, and genetic counselling. Mol. Syndromol. 2018, 9, 60–69.
- Gunel, M.; Laurans, M.S.; Shin, D.; DiLuna, M.L.; Voorhees, J.; Choate, K.; Nelson-Williams, C.; Lifton, R.P. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc. Natl. Acad. Sci. USA 2002, 99, 10677–10682.
- Fischer, A.; Zalvide, J.; Faurobert, E.; Albiges-Rizo, C.; Tournier-Lasserve, E. Cerebral cavernous malformations: From CCM genes to endothelial cell homeostasis. Trends Mol. Med. 2013, 19, 302–308.
- Dashti, S.R.; Hoffer, A.; Hu, Y.C.; Selman, W.R. Molecular genetics of familial cerebral cavernous malformations. Neurosurg. Focus 2006, 21, 2.
- McDonald, D.A.; Shi, C.; Shenkar, R.; Gallione, C.J.; Akers, A.L.; Li, S.; De Castro, N.; Berg, M.J.; Corcoran, D.L.; Awad, I.A.; et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: Evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 2014, 23, 4357–4370.
- Kar, S.; Samii, A.; Bertalanffy, H. PTEN/PI3K/Akt/VEGF signaling and the cross talk to KRIT1, CCM2, and PDCD10 proteins in cerebral cavernous malformations. Neurosurg. Rev. 2015, 38, 229–236.
- Plummer, N.W.; Zawistowski, J.S.; Marchuk, D.A. Genetics of cerebral cavernous malformations. Curr. Neurol. Neurosci. Rep. 2005, 5, 391–396.
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillip, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207.
- Wang, K.; Zhou, H.J.; Wang, M. CCM3 and cerebral cavernous malformation disease. Stroke Vasc. Neurol. 2019, 4, 67–70.
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2005, 76, 42–51.
- Denier, C.; Labauge, P.; Bergametti, F.; Marchelli, F.; Riant, F.; Arnoult, M.; Maciazek, J.; Vicaut, E.; Brunereau, L.; Tournier-Lasserve, E.; et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 2006, 60, 550–5566.
- Chan, A.C.; Li, D.L.; Berg, M.J.; Whitehead, K.J. Recent insights into cerebral cavernous malformations: Animal models of CCM and the human phenotype. FEBS J. 2010, 277, 1076–1083.
- Riant, F.; Bergametti, F.; Ayrignac, X.; Boulday, G.; Tournier-Lasserve, E. Recent insights into cerebral cavernous malformations: The molecular genetics of CCM. FEBS J. 2010, 277, 1070–1075.
- Denier, C.; Gasc, J.M.; Chapon, F.; Domenga, V.; Lescoat, C.; Joutel, A.; Tournier-Lasserve, E. Krit1/cerebral cavernous malformation 1 mRNA is preferentially expressed in neurons and epithelial cells in embryo and adult. Mech. Dev. 2002, 117, 363–367.
- Whitehead, K.J.; Plummer, N.W.; Adams, J.A.; Marchuk, D.A.; Li, D.Y. Ccm1 is required for arterial morphogenesis: Implications for the etiology of human cavernous malformations. Development 2004, 131, 1437–1448.
- Zhang, J.; Clatterbuck, R.E.; Rigamonti, D.; Dietz, H.C. Mutations in KRIT1 in familial cerebral cavernous malformations. Neurosurgery 2000, 46, 1272–1277.
- Choquet, H.; Pawlikowska, L.; Lawton, M.T.; Kim, H. Genetics of cerebral cavernous malformations: Current status and future prospects. J. Neurosurg. Sci. 2015, 59, 211–220.
- Cavé-Riant, F.; Denier, C.; Labauge, P.; Cécillon, M.; Maciazek, J.; Joutel, A.; Laberge-Le Couteulx, S.; Tournier-Lasserve, E. Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with cerebral cavernous malformations. Eur. J. Hum. Genet. 2002, 10, 733–740.
- Gault, J.; Sain, S.; Hu, L.J.; Awad, I.A. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery 2006, 59, 1278–1284.
- Battistini, S.; Rocchi, R.; Cerase, A.; Citterio, A.; Tassi, L.; Lando, G.; Patrosso, M.C.; Galli, R.; Brunori, P.; Sgrò, D.L.; et al. Clinical, magnetic resonance imaging, and genetic study of 5 Italian families with cerebral cavernous malformation. Arch. Neurol. 2007, 64, 843–848.
- Akers, A.L.; Johnson, E.; Steinberg, G.K.; Zabramski, J.M.; Marchuk, D.A. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. 2009, 18, 919–930.
- Ricci, C.; Cesare, A.; Riolo, G.; Manasse, G.; Battistini, S. KRIT1 gene in patients with cerebral cavernous malformations: Clinical features and molecular characterization of novel variants. J. Mol. Neurosci. 2021. (accepted).
- Ricci, C.; Riolo, G.; Battistini, S. Molecular genetic analysis of Cerebral Cavernous Malformations (CCM): An update. Vessel Plus. (under review).
- Denier, C.; Goutagny, S.; Labauge, P.; Krivosic, V.; Arnoult, M.; Cousin, A.; Benabid, A.L.; Comoy, J.; Frerebeau, P.; Gilbert, B.; et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2004, 74, 326–337.
- De Vos, I.J.; Vreeburg, M.; Koek, G.H.; van Steensel, M.A. Review of familial cerebral cavernous malformations and report of seven additional families. Am. J. Med. Genet. A 2017, 173, 338–351.
- Liu, W.; Draheim, K.M.; Zhang, R.; Calderwood, D.A.; Boggon, T.J. Mechanism for KRIT1 release of ICAP1-mediated integrin activation suppression. Mol. Cell. 2013, 49, 719–729.
- Hogan, B.M.; Bussmann, J.; Wolburg, H.; Schulte-Merker, S. CCM1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum. Mol. Genet. 2008, 17, 2424–2432.
- Boulday, G.; Blécon, A.; Petit, N.; Chareyre, F.; Garcia, L.A.; Niwa-Kawakita, M.; Giovannini, M.; Tournier-Lasserve, E. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: Implications for human cerebral cavernous malformations. Dis. Models Mech. 2009, 2, 168–177.
- Shenkar, R.; Venkatasubramanian, P.N.; Wyrwicz, A.M.; Zhao, J.C.; Shi, C.; Akers, A.; Marchuk, D.A.; Awad, I.A. Advanced magnetic resonance imaging of cerebral cavernous malformations: Part II. Imaging of lesions in murine models. Neurosurgery 2008, 63, 790–797.
- Haasdijk, R.; Cheng, C.; Maat-Kievit, A.; Duckers, H.J. Cerebral cavernous malformations: From molecular pathogenesis to genetic counselling and clinical management. Eur. J. Hum. Genet. 2012, 20, 134–140.
- Yang, L.; Wu, J.; Zhang, J. A novel CCM2 gene mutation associated with cerebral cavernous malformation. Front. Neurol. 2020, 11, 70.
- He, Y.; Zhang, H.; Yu, L.; Gunel, M.; Boggon, T.J.; Chen, H.; Min, W. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci. Signal. 2010, 3, 26.
- Gianfrancesco, F.; Esposito, T.; Penco, S.; Maglione, V.; Liquori, C.L.; Patrosso, M.C.; Zuffardi, O.; Ciccodicola, A.; Marchuk, D.A.; Squitieri, F. ZPLD1 gene is disrupted in a patient with balanced translocation that exhibits cerebral cavernous malformations. Neuroscience 2008, 155, 345–349.
- Cavalcanti, D.D.; Kalani, M.Y.; Martirosyan, N.L.; Eales, J.; Spetzler, R.F.; Preul, M.C. Cerebral cavernous malformations: From genes to proteins to disease. J. Neurosurg. 2012, 116, 122–132.
- Abou-Fadel, J.; Qu, Y.; Gonzalez, E.M.; Smith, M.; Zhang, J. Emerging roles of CCM genes during tumorigenesis with potential application as novel biomarkers across major types of cancers. Oncol. Rep. 2020, 43, 1945–1963.
- Su, V.L.; Calderwood, D.A. Signalling through cerebral cavernous malformation protein networks. Open Biol. 2020, 10, 200263.
- Orsenigo, F.; Conze, L.L.; Jauhiainen, S.; Corada, M.; Lazzaroni, F.; Malinverno, M.; Sundell, V.; Cunha, S.I.; Brännström, J.; Globisch, M.A.; et al. Mapping endothelial-cell diversity in cerebral cavernous malformations at single-cell resolution. eLife 2020, 9, e61413.
- Yadla, S.; Jabbour, P.M.; Shenkar, R.; Shi, C.; Campbell, P.G.; Awad, I.A. Cerebral cavernous malformations as a disease of vascular permeability: From bench to bedside with caution. Neurosurg. Focus 2010, 29, 4.
- Draheim, K.M.; Fisher, O.S.; Boggon, T.J.; Calderwood, D.A. Cerebral cavernous malformation proteins at a glance. J. Cell. Sci. 2014, 127, 701–707.
- Fisher, O.S.; Boggon, T.J. Signaling pathways and the cerebral cavernous malformations proteins: Lessons from structural biology. Cell. Mol. Life Sci. 2014, 71, 1881–1892.
- Serebriiskii, I.; Estojak, J.; Sonoda, G.; Testa, J.R.; Golemis, E.A. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 1997, 15, 1043–1049.
- Li, X.; Zhang, R.; Draheim, K.M.; Liu, W.; Calderwood, D.A.; Bogon, T.J. Structural basis for small G protein effector interaction of Ras-related protein 1 (Rap1) and adaptor protein Krev interaction trapped 1 (KRIT1). J. Biol. Chem. 2012, 287, 22317–22327.
- Gingras, A.R.; Puzon-McLaughlin, W.; Ginsberg, M.H. The structure of the ternary complex of krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the heart of glass (HEG1) cytoplasmic tail. J. Biol. Chem. 2013, 288, 23639–23649.
- Gingras, A.R.; Liu, J.J.; Ginsberg, M.H. Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell. Biol. 2012, 199, 39–48.
- Glading, A.; Han, J.; Stockton, R.A.; Ginsberg, M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell. Biol. 2007, 179, 247–254.
- Van den Berg, M.C.W.; Burgering, B.M.T. CCM1 and the second life of proteins in adhesion complexes. Cell. Adh. Migr. 2014, 8, 146–157.
- Draheim, K.M.; Huet-Calderwood, C.; Simon, B.; Calderwood, D.A. Nuclear localization of Integrin Cytoplasmic Domain-associated Protein-1 (ICAP1) influences β1 integrin activation and recruits krev interaction trapped-1 (KRIT1) to the nucleus. J. Biol. Chem. 2017, 292, 1884–1898.
- Zawistowski, J.S.; Serebriiskii, I.G.; Lee, M.F.; Golemis, E.A.; Marchuk, D.A. KRIT1 association with the integrin-binding protein ICAP-1: A new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 2002, 11, 389–396.
- Marchuk, D.A.; Srinivasan, S.; Squire, T.L.; Zawistowski, J.S. Vascular morphogenesis: Tales of two syndromes. Hum. Mol. Genet. 2003, 12, 97–112.
- Seker, A.; Pricola, K.L.; Guclu, B.; Ozturk, A.K.; Louvi, A.; Gunel, M. CCM2 expression parallels that of CCM1. Stroke 2006, 37, 518–523.
- Zawistowski, J.S.; Stalheim, L.; Uhlik, M.T.; Abell, A.N.; Ancrile, B.B.; Johnson, G.L.; Marchuk, D.A. CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 2005, 14, 2521–2531.
- Stiegler, A.L.; Zhang, R.; Liu, W.; Boggon, T.J. Structural determinants for binding of sorting nexin 17 (SNX17) to the cytoplasmic adaptor protein Krev interaction trapped 1 (KRIT1). J. Biol. Chem. 2014, 289, 25362–25373.
- Béraud-Dufour, S.; Gautier, R.; Albiges-Rizo, C.; Chardin, P.; Faurobert, E. Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 2007, 274, 5518–5532.
- Francalanci, F.; Avolio, M.; De Luca, E.; Longo, D.; Menchise, V.; Guazzi, P.; Sgrò, F.; Marino, M.; Goitre, L.; Balzac, F.; et al. Structural and functional differences between KRIT1A and KRIT1B isoforms: A framework for understanding CCM pathogenesis. Exp. Cell. Res. 2009, 315, 285–303.
- Sedgwick, S.G.; Smerdon, S.J. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 1999, 24, 311–316.
- Hilder, T.L.; Malone, M.H.; Bencharit, S.; Colicelli, J.; Haystead, T.A.; Johnson, G.L.; Wu, C.C. Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 2007, 6, 4343–4355.
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896.
- Kleaveland, B.; Zheng, X.; Liu, J.J.; Blum, Y.; Tung, J.J.; Zou, Z.; Sweeney, S.M.; Chen, M.; Guo, L.; Lu, M.M.; et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 2009, 15, 584.
- Rosen, J.N.; Sogah, V.M.; Ye, L.Y.; Mably, J.D. CCM2-like is required for cardiovascular development as a novel component of the Heg-CCM pathway. Dev. Biol. 2013, 376, 74–85.
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184.
- Wei, S.; Li, Y.; Polster, S.P.; Weber, C.R.; Awad, I.A.; Shen, L. Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation. Int. J. Mol. Sci. 2020, 21, 675.
- Zheng, X.; Xu, C.; Smith, A.O.; Stratman, A.N.; Zou, Z.; Kleaveland, B.; Yuan, L.; Didiku, C.; Sen, A.; Liu, X.; et al. Dynamic regulation of the cerebral cavernous malformation pathway controls vascular stability and growth. Dev. Cell. 2012, 23, 342–355.
- Cullere, X.; Plovie, E.; Bennett, P.M.; MacRae, C.A.; Mayadas, T.N. The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proc. Natl. Acad. Sci. USA 2015, 112, 14284–14289.
- Ding, J.; Wang, X.; Li, D.F.; Hu, Y.; Zhang, Y.; Wang, D.C. Crystal structure of human programmed cell death 10 complexed with inositol-(1,3,4,5)-tetrakisphosphate: A novel adaptor protein involved in human cerebral cavernous malformation. Biochem. Biophys. Res. Commun. 2010, 399, 587–592.
- Draheim, K.M.; Li, X.; Zhang, R.; Fisher, O.S.; Villari, G.; Boggon, T.J.; Calderwood, D.A. CCM2-CCM3 interaction stabilizes their protein expression and permits endothelial network formation. J. Cell. Biol. 2015, 208, 987–1001.
- Goudreault, M.; D’Ambrosio, L.M.; Kean, M.J.; Mullin, M.J.; Larsen, B.G.; Sanchez, A.; Chaudhry, S.; Chen, G.I.; Sicheri, F.; Nesvizhskii, A.I.; et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol. Cell. Proteom. 2009, 8, 157–171.
- Ceccarelli, D.F.; Laister, R.C.; Mulligan, V.K.; Kean, M.J.; Goudreault, M.; Scott, I.C.; Derry, W.B.; Chakrabartty, A.; Gingras, A.C.; Sicheri, F. CCM3/PDCD10 heterodimerizes with germinal center kinase III (GCKIII) proteins using a mechanism analogous to CCM3 homodimerization. J. Biol. Chem. 2011, 286, 25056–25064.
- Padarti, A.; Zhang, J. Recent advances in cerebral cavernous malformation research. Vessel Plus 2018, 2, 21.
- Fidalgo, M.; Fraile, M.; Pires, A.; Force, T.; Pombo, C.; Zalvide, J. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J. Cell. Sci. 2010, 123, 1274–1284.
- Kean, M.J.; Ceccarelli, D.F.; Goudreault, M.; Sanches, M.; Tate, S.; Larsen, B.; Gibson, L.C.; Derry, W.B.; Scott, I.C.; Pelletier, L.; et al. Structure-function analysis of core STRIPAK Proteins: A signaling complex implicated in Golgi polarization. J. Biol. Chem. 2011, 286, 25065–25075.
- Jahn, R.; Südhof, T.C. Membrane fusion and exocytosis. Annu. Rev. Biochem. 1999, 68, 863–911.
- Zhao, H.; Mleynek, T.M.; Li, D.Y. Dysregulated exocytosis of angiopoietin-2 drives cerebral cavernous malformation. Nat. Med. 2016, 22, 971–973.
- Zhou, H.J.; Qin, L.; Zhang, H.; Tang, W.; Ji, W.; He, Y.; Liang, X.; Wang, Z.; Yuan, Q.; Vortmeyer, A.; et al. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat. Med. 2016, 22, 1033–1042.
- Peng, W.; Wu, X.; Feng, D.; Zhang, Y.; Chen, X.; Ma, C.; Shen, H.; Li, X.; Li, H.; Zhang, J.; et al. Cerebral cavernous malformation 3 relieves subarachnoid hemorrhage-induced neuroinflammation in rats through inhibiting NF-kB signaling pathway. Brain Res. Bull. 2020, 160, 74–84.
- Dibble, C.F.; Horst, J.A.; Malone, M.H.; Park, K.; Temple, B.; Cheeseman, H.; Barbaro, J.R.; Johnson, G.L.; Bencharit, S. Defining the functional domain of programmed cell death 10 through its interactions with phosphatidylinositol-3,4,5-trisphosphate. PLoS ONE 2010, 5, 11740.
- Mardakheh, F.K.; Self, A.; Marshall, C.J. Rho binding to FAM65A regulates Golgi reorientation during cell migration. J. Cell. Sci. 2016, 129, 4466–4479.
- Harel, L.; Costa, B.; Tcherpakov, M.; Zapatka, M.; Oberthuer, A.; Hansford, L.M.; Vojvodic, M.; Levy, Z.; Chen, Z.Y.; Lee, F.S.; et al. CCM2 mediates death signaling by the TrkA receptor tyrosine kinase. Neuron 2009, 63, 585–591.
- Guclu, B.; Ozturk, A.K.; Pricola, K.L.; Bilguvar, K.; Shin, D.; O’Roak, B.J.; Gunel, M. Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery 2005, 57, 1008–1013.
- Fidalgo, M.; Guerrero, A.; Fraile, M.; Iglesias, C.; Pombo, C.M.; Zalvide, J. Adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins ezrin/radixin/moesin by mammalian Ste20-4 to protect cells from oxidative stress. J. Biol. Chem. 2012, 287, 11556–11565.
- Baranoski, J.F.; Kalani, M.Y.; Przybylowski, C.J.; Zabramski, J.M. Cerebral cavernous malformations: Review of the genetic and protein–protein interactions resulting in disease pathogenesis. Front. Surg. 2016, 3, 60.



