 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lakmie Gunarathne | + 3261 word(s) | 3261 | 2021-02-19 03:47:46 | | | |
| 2 | Vivi Li | Meta information modification | 3261 | 2021-03-03 07:42:09 | | |
Video Upload Options
There is considerable experimental evidence that the renin angiotensin system (RAS) plays a central role in both hepatic fibrogenesis and portal hypertension. Angiotensin converting enzyme (ACE), a key enzyme of the classical RAS, converts angiotensin I (Ang I) to angiotensin II (Ang II), which acts via the Ang II type 1 receptor (AT1R) to stimulate hepatic fibrosis and increase intrahepatic vascular tone and portal pressure. Inhibitors of the classical RAS, drugs which are widely used in clinical practice in patients with hypertension, have been shown to inhibit liver fibrosis in animal models but their efficacy in human liver disease is yet to be tested in adequately powered clinical trials. Small trials in cirrhotic patients have demonstrated that these drugs may lower portal pressure but produce off-target complications such as systemic hypotension and renal failure. More recently, the alternate RAS, comprising its key enzyme, ACE2, the effector peptide angiotensin-(1–7) (Ang-(1–7)) which mediates its effects via the putative receptor Mas (MasR), and Mas related G protein-coupled receptor type-D (MrgD) has also been implicated in the pathogenesis of liver fibrosis and portal hypertension. This system is activated in both preclinical animal models and human chronic liver disease and it is now well established that the alternate RAS counter-regulates many of the deleterious effects of the ACE-dependent classical RAS. Work from our laboratory has demonstrated that liver-specific ACE2 overexpression reduces hepatic fibrosis and liver perfusion pressure without producing off-target effects. On the other hand, activation of the alternate RAS aggravates portal hypertension in cirrhosis via increasing splanchnic vasodilatation and elevating portal blood flow into the liver. Recent studies suggest that the blockers of the receptors of alternate RAS, such as the MasR and MrgD, increase splanchnic vascular resistance in cirrhotic animals, and thus drugs targeting the alternate RAS may be useful in the treatment of portal hypertension.
1. Introduction
Fibrosis or scarring of the liver is initiated as a part of the wound healing response to tissue injury. The end result of chronic fibrotic injury to the liver is cirrhosis, in which there is extensive scar formation, distortion of liver parenchyma by septae and nodule formation, and alterations in blood flow and this can finally lead to liver failure [1][2]. A major outcome of cirrhosis is the development of portal hypertension which is responsible for many of the complications including life-threatening variceal bleeding [3]. Cirrhosis has become the 11th most common cause for deaths in humans and was responsible for approximately 1.2 million deaths worldwide in 2016 [4]. The most common causes for chronic liver disease (CLD) include chronic viral infections (e.g., hepatitis B and C), excessive alcohol consumption, non-alcoholic fatty liver disease (NAFLD) and cholestatic diseases such as primary biliary cholangitis and primary sclerosing cholangitis [2][5][6].
Despite being a major health problem, there is no specific medical treatment for cirrhosis. Therefore, treatments that target the causative factors and managing complications associated with cirrhosis including portal hypertension are the only currently available options. Thus, if the causative agent is viral hepatitis C (Hep C), treatment with antiviral therapies leads to cessation and even reversal of tissue fibrosis [7]. Treatment options for established cirrhosis and portal hypertension are limited with the major therapy being non-selective beta-blockers (NSBB). Recent research shows that the renin angiotensin system (RAS) is activated during the development of cirrhosis and contributes to the pathogenesis of both liver fibrosis and portal hypertension [3][6].
2. The Renin Angiotensin System (RAS)
In normal physiology, the RAS plays a very important role in vascular resistance and regulation of blood pressure, sodium and water homeostasis, and tissue remodeling during a tissue injury. The RAS comprises two arms known as the “classical arm” and the “alternate arm” (protective arm). The alternate arm of the RAS plays a major role in counter-balancing many of the deleterious effects of the classical RAS [8].
2.1. The Classical Arm of the Renin Angiotensin System
The classical arm of the RAS can be thought of as a linear cascade where angiotensinogen is converted to the effector peptide of the system, angiotensin II (Ang II). Angiotensinogen is produced mainly in the liver by hepatocytes and released into the circulation. Circulating angiotensinogen is converted to the decapeptide angiotensin I (Ang I) by renin, an enzyme produced by the juxtaglomerular apparatus of the kidney. Thereafter, angiotensin converting enzyme (ACE), mainly present in the lungs, converts Ang I to Ang II [9]. There is also an ACE independent pathway of Ang II production from Ang I, which is regulated via a serine endopeptidases named chymase [10][11]. Ang II mediates its effects through two G-protein-coupled receptors, Ang II type 1 receptor (AT1R) and Ang II type 2 receptor (AT2R). AT1R is the predominant receptor type during adult life whereas AT2R, although having some functional role in adults, has been postulated to function mainly during fetal life [12].
Ang II by binding to the AT1R mediates classical RAS functions which include vasoconstriction (by directly affecting vascular smooth muscle cells), sodium homeostasis (by increasing sodium reabsorption through renal tubules and by stimulating the adrenal gland to release aldosterone), increasing thirst (by acting on AT1R in the brain), induction of inflammation and the wound healing response via secretion of cytokines, chemokines, and extracellular matrix proteins. Ang II also acts as a prooxidant and prothrombotic agent and interferes at several steps of intracellular insulin signaling pathways such as the PI3 kinase (phosphoinositide 3-kinase) and MAP (mitogen activated protein kinase) kinase pathways [13][14].
2.2. The Alternate Arm of the Renin Angiotensin System
Whilst the physiological role of the classical RAS is well-established, the discovery of a new RAS enzyme, ACE2, a homologue of ACE, has dramatically changed our understanding of the RAS physiology [15][16]. The ‘alternate or the protective arm’ of the RAS driven by ACE2 is considered as the counter-regulatory arm of the classical RAS [3].
In the late 80s, researchers discovered a biologically active heptapeptide angiotensin-(1–7) (Ang-(1–7)) of the RAS [17]. The enzymatic pathway responsible for producing Ang-(1–7) came to light when ACE2 was discovered in the year 2000 by two independent laboratories [15][16]. It is now known that Ang-(1–7) peptide is produced by ACE2 after cleavage of a carboxyl terminal single amino acid from the 8-amino acid peptide Ang II. ACE2 is a zinc-metalloproteinase and a type-1 transmembrane protein which consists of 805 amino acids with a single transmembrane alpha-helical portion, an extracellular N-terminus portion containing the catalytically active domain and an internal inactive C-terminus [9]. ACE2 is structurally similar to ACE; however, it is functionally different with different substrate affinities than that of ACE, and ACE2 is resistant to ACE inhibitors (ACEi). The major action of ACE2 is to break down Ang II to Ang-(1–7), which acts through the Mas receptor (MasR), a G protein-coupled receptor (GPCR). Ang (1–7) is subsequently metabolized via ACE into Ang-(1–5) and via the other neutral endopeptidases (NEP), neprilysin into Ang-(1–4) [18]. The ACE2/Ang-(1–7)/MasR arm counter-regulates many of the actions of the classical RAS, thus producing opposing effects to those of the classical RAS, including antihypertensive, anti-inflammatory, antithrombotic, antiproliferative, and antifibrotic effects (Figure 1).
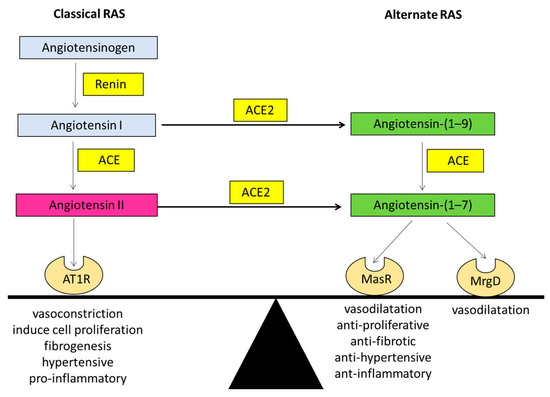
Figure 1. The ‘balance’ between the two arms of the renin angiotensin system (RAS). Graphical representation of the classical arm (ACE/Angiotensin II/AT1R) and the alternate arm (ACE2/Angiotensin-(1–7)/MasR) of the RAS where the alternate arm counter-balances the deleterious effects of the classical arm. Angiotensin II can exert its effects via the angiotensin II type 1 receptor (AT1R). Whilst angiotensin-(1–7) of the alternate RAS acts mainly via the Mas receptor (MasR), recent evidence suggests that it also transduces its signal via the Mas related G protein-coupled receptor type-D (MrgD). ACE: angiotensin converting enzyme; ACE2: angiotensin converting enzyme 2.
Although the MasR is recognized to be the functional receptor for Ang-(1–7) [19], Santos and colleagues in 2003 suggested that certain effects of Ang-(1–7) may not be mediated through MasR [20]. This was supported by the finding that MasR blocker D-Ala7-Ang-(1–7) (A779) could not block the Ang-(1–7) mediated vasodilatation in the rat aorta, however, these effects were completely blocked by the Ang-(1–7) antagonist D-Pro7-Ang-(1–7) (D-Pro) [21]. Subsequently, a study by Gembardt and colleagues showed that upon stimulation with Ang-(1–7), COS cells overexpressing MasR or a newly characterized Ang-(1–7) receptor, the Mas related G protein-coupled receptor type-D (MrgD), released arachidonic acid [22]. Lautner and colleagues later showed that in addition to Ang-(1–7), the RAS peptide alamandine also activates the MrgD, and the vasodilatory effects produced by alamandine were blocked by the MrgD blocker D-Pro [23]. Work from our laboratory provided further evidence by showing that D-Pro blocked Ang-(1–7) mediated vasodilatation in perfused cirrhotic rat livers whilst the MasR blocker A779 had no effect [24].
In a recent study, it was shown that when transfected with either the MasR or MrgD, mesangial cells release cyclic adenosine monophosphate (cAMP) in the presence of Ang-(1–7) [25]. Moreover, in MasR or MrgD transfected HEK293 cells, blockade of MasR and MrgD with A779 and D-Pro, respectively, abolished the release of cAMP. Moreover, the effects of MrgD stimulation in-vitro were confirmed by in-vivo functional studies by showing that the hemodynamic responses to a bolus injection of Ang-(1–7) were blunted in MrgD knockout (MrgD-KO) mice compared with the controls. With these findings, it is now considered that the alternate arm of the RAS consists of ACE2/Ang-(1–7)/MasR/MrgD [25].
3. The Role of Classical RAS in Liver Fibrosis
Traditionally, the RAS was considered as an endocrine system. However, recent studies have shown that there is a local RAS which functions in an autocrine and/or paracrine manner, in major organs such as heart, the kidneys and liver [26][27]. The local RAS is activated in response to a tissue injury, which leads to a wound healing response with upregulated expression of the RAS components at the site of repair [28]. Initially it was discovered that the classical RAS plays an important role in tissue repair and organ fibrosis in heart disease and chronic kidney disease (CKD), and RAS inhibitors have been shown to have benefits in these conditions beyond those that are due to their antihypertensive effects [29][30].
There is also substantial evidence that Ang II is a major mediator in hepatic fibrosis. Serum Ang II concentration is significantly elevated in patients with cirrhosis [31][32] and the local RAS in the liver is activated as a response to the chronic injury. Findings from our laboratory and others showed that the hepatic expression of the classical RAS comprising ACE, Ang II and AT1R is increased in the diseased liver compared to those in healthy livers [3][33][34][35][36][37]. Moreover, these studies reported that the expression of the RAS components was localized to the areas of active fibrogenesis, suggesting that the local classic RAS plays a major role during the progression of liver fibrosis.
Following liver injury a major cell type responsible for the wound healing response is the hepatic stellate cell (HSC) [38]. Ang II plays a major role in the activation and phenotypic transformation of HSCs into active myofibroblasts which drive tissue fibrosis. Whilst quiescent HSCs have minimal expression of the RAS components and do not produce Ang II, activated HSCs express all components of RAS including angiotensinogen, renin, ACE, and AT1R [28]. Activated HSCs thus have the potential to synthesize Ang II, which acts on AT1R in an autocrine fashion [28][39][40], stimulating their proliferation [41]. The molecular mechanism responsible for HSC activation appears to be the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, an enzyme which produces reactive oxygen species (ROS) in response to Ang II. Inhibition of NADPH oxidase with diphenylene iodonium (DPI) blocked Ang II-induced ROS production in cultured HSCs, confirming that NADPH oxidase mediates the increase in ROS after Ang II stimulation [31]. This evidence suggests that Ang II has the potential to induce HSCs to produce ROS that can further stimulate the fibrogenic process in an autocrine and/or paracrine manner. In addition, Ang II acts as a powerful chemoattractant for activated HSCs [31]. Thus, Ang II becomes a contributing factor for HSC proliferation, activation and accumulation at the site of injury.
Furthermore, Ang II exerts a direct influence on endothelial function [13]. In general, normal endothelial function is determined by the cell redox state, which is controlled by a homeostatic balance between nitric oxide and ROS. Griendling and colleagues in 1994 showed that prolonged exposure of vascular smooth muscle cells (VSMCs) to Ang II induced oxidative stress and ROS generation due to activation of NADPH, the enzyme responsible for intracellular superoxide generation. In addition, it has been shown that Ang II stimulates the release of endothelin-1, a potent vasoconstrictor and inducer of smooth muscle cell proliferation in vasculature, while altering the balance between the fibrinolysis and coagulation processes [13].
When normal liver sinusoidal endothelial function is disturbed by factors such as Ang II induced ROS generation, the damaged liver sinusoidal endothelial cells (LSEC) are replaced by bone marrow (BM) derived sinusoidal endothelial cell progenitor cells (sprocs) in order to compensate for the loss. These BM sprocs which lack LSEC fenestrae arrange along the sinusoids and an organized basement membrane is laid down. This process of dedifferentiation of injured LSECs leads to “capillarization” of the sinusoids [42]. Although normal LSECs are responsible for the production of heparin-binding epidermal growth factor (HB-EGF), which maintains HSC quiescence (Figure 2), the immature LSECs derived from BM sprocs have less or reduced capacity to release HB-EGF which leads to activation of HSCs [42][43]. In addition, dedifferentiated LSECs express EIIIA fibronectin isoform which influences quiescence HSCs to change their phenotype to the activated form [44][45]. Thus, LSECs perform a critical crosstalk function in the HSC activation process in hepatic fibrosis by simultaneous reduction in HB-EGF and expression of EIIIA (Figure 2).
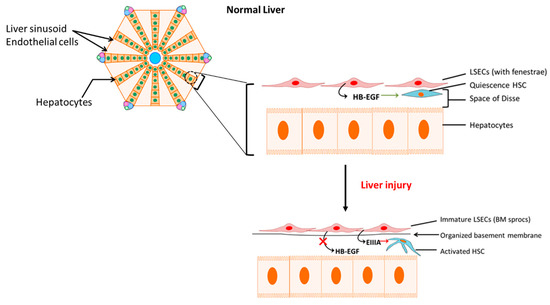
Figure 2. Activation of hepatic stellate cells (HSCs) by immature liver sinusoidal endothelial cells (LSECs). Reduced release of heparin-binding epidermal growth factor (HB-EGF) with concomitant increase in fibronectin isoform EIIIA release by bone marrow-derived immature LSECs activates HSCs. BM sprocs, bone marrow (BM) derived sinusoidal endothelial cell progenitor cells (sprocs). BM: bone marrow; HB-EGF: heparin-binding epidermal growth factor.
ACE Inhibitors (ACEi) and Angiotensin II Type 1 Receptor Blockers (ARBs) in Liver Fibrosis
Since Ang II plays a major role in liver fibrosis, ACEi and ARBs have been studied as potential antifibrotic therapies in liver disease [40]. ACE inhibitors are widely used in patients with high blood pressure and chronic heart failure. Studies in preclinical models suggest that ACEi attenuate the expression of transforming growth factor beta 1 (TGF-β1), collagen and other extra cellular matrix (ECM) proteins including matrix metalloproteinase (MMP)-2 and MMP-9, leading to attenuation of fibrosis [13][46][47].
There have also been several animal studies which have examined the effects of ARBs in liver diseases. Studies in bile duct ligated (BDL) rats have shown that ARB, telmisartan decreased gene expression of ACE, AT1R, collagen type III, and TGF-β1 while increasing the expression of ACE2 and MasR with concomitant reduction in hepatic fibrosis [48][49]. Another ARB, losartan, significantly ameliorated the progression of hepatic fibrosis induced by carbon tetrachloride (CCl4) in rats along with significant reductions in gene expression levels of AT1R and TGF-β1 [50]. Moreover, a study performed in rats with NASH induced by feeding methionine-choline deficient (MCD) diet showed that another ARB, olmesartan, attenuated serum aspartate transaminase (AST) levels, HSC activity, oxidative stress, gene expression of TGF-β1 and collagen, collectively leading to improved liver fibrosis likely resulting from reduced activation of HSCs [51].
Despite evidence from animal studies there is a relative lack of clinical studies of RAS inhibition in human liver fibrosis. A retrospective study carried out by Corey and colleagues suggested a reduction in liver fibrosis and necroinflammation in patients with chronic hepatitis C who received ACE inhibitors and ARBs as a treatment for hypertension [52]. The study reviewed 284 liver biopsies from the patients who were evaluated at an outpatient hepatology clinic during a 5 year period. The patients were classified into three groups: Group 1—hypertensive patients (n = 143) who received ACE inhibitors (captopril, enalapril, lisinopril, quinapril, and trandolapril) or ARBs (losartan, valsartan and irbesartan); Group 2—hypertensive patients (n = 91) treated with β-adrenergic antagonists, calcium channel antagonists, diuretics, α-adrenergic antagonists, and vasodilators; Group 3—patients (n = 50) with chronic hepatitis C infection who did not have hypertension. The results showed a significantly low mean Ishak fibrosis score in patients included in the Group 1 who received ACE inhibitors and ARBs compared to the patients in Group 2 who were treated with other treatments for hypertension. The patients with no hypertension included in the Group 3 showed the lowest mean fibrosis score supporting the theory that hypertension and activation of RAS contributes to fibrosis progression. Another register-based cohort study of patients (n = 70,546) with a first-time diagnosis of chronic liver disease between 2005 and 2012 in Sweden revealed a marked reduction in liver-related mortality among patients with alcoholic liver disease who received ACE inhibitors [53].
Only a small number of randomized studies have evaluated the effects of RAS blockade in liver fibrosis. A randomized open-label controlled study performed by a group of researchers from South Korea in 2012 investigated the antifibrotic effect of ARBs in patients with compensated alcoholic liver disease [54]. They treated 42 patients with candesartan and ursodeoxycholic acid (UDCA), and the control group (n = 43) received UDCA alone for 6 months. There was a reduction in Laennec fibrosis score, area of fibrosis, hydroxyproline level, and α-smooth muscle actin in the candesartan treated group. Another randomized clinical trial performed in 2011 in a group of selected patients with cirrhosis compared those who received (n = 24) and those who did not receive (n = 24) olmesartan treatment for one year. There was a reduction in TGF-β1 in the olmesartan treated group but not in hepatic fibrosis markers which include serum hyaluronic acid, type IV collagen, and procollagen III N-terminal propeptide levels [55]. In 2007, Debernardi-Venon and colleagues from Italy published data from a randomized clinical trial of candesartan treatment conducted in 47 compensated Child A and Child B cirrhotic patients. The results showed a significant reduction in serum fibrosis marker hyaluronic acid in candesartan treated patients compared to the untreated patients [56]. Although there is a reduction in hyaluronic acid level in treated patients, the results were not supported by histological data. Between 2004 and 2006, a 48-month follow up study was conducted on 89 patients with cirrhosis associated with hepatocellular carcinoma (HCC) in Japan using combination therapy with branched-chain amino acid (BCAA) granules and an ACE inhibitor, perindopril [57]. The serum fibrosis markers hyaluronic acid and type IV collagen 7S were measured in groups of patients who received a combination therapy with BCAA granules and perindopril and two single-treatment groups who received either perindopril or BCAA and compared to a control group. The patients included into this study were confirmed to be free of any residual HCC, alcohol consumption and also the status of insulin resistance (IR). The results of this study show that combination therapy of BCAA and perindopril improved serum fibrosis markers as compared to either ACE inhibitor or BCAA alone. Another randomized study conducted in 30 patients with early stages of chronic hepatitis C in Japan showed that oral losartan and UDCA administration has the potential to reduce serum type IV collagen and TGF-β1 compared to UDCA alone, but not liver fibrosis as indicated by METAVIR fibrosis score [58]. A recent double-blind randomized-controlled trial which was designed to study the efficacy of losartan to reduce or reverse the progression of fibrosis in patients with nonalcoholic steatohepatitis (NASH) was unable to show a positive outcome due to the widespread use of ACE inhibitors and ARBs in patients with NASH [59].
Thus, there are some studies which suggest beneficial effects of ACE inhibitors and ARBs as antifibrotic agents, [52][53][57]; however, the evidence is conflicting and there is a definite need for further large randomized placebo controlled clinical trials [60]. In addition, it should be noted that there are also concerns about the use of RAS inhibitors in advanced cirrhosis [61][62] as they can induce arterial hypotension and renal function impairment.
References
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669.
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851.
- Grace, J.A.; Herath, C.B.; Mak, K.Y.; Burrell, L.M.; Angus, P.W. Update on new aspects of the renin–angiotensin system in liver disease: Clinical implications and new therapeutic options. Clin. Sci. 2012, 123, 225–239.
- WHO. Summary Tables of Mortality Estimates by Cause, Age and Sex, Globally and by Region, 2000–2016; WHO: Geneva, Switzerland, 2019.
- Zhou, W.-C.; Zhang, Q.-B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324.
- Rajapaksha, I.G.; Angus, P.W.; Herath, C.B. Current therapies and novel approaches for biliary diseases. World J. Gastrointest. Pathophysiol. 2019, 10, 1–10.
- Berzigotti, A. Advances and challenges in cirrhosis and portal hypertension. BMC Med. 2017, 15, 200.
- Warner, F.J.; Lubel, J.S.; McCaughan, G.W.; Angus, P.W. Liver fibrosis: A balance of ACEs? Clin. Sci. 2007, 113, 109–118.
- Lubel, J.S.; Herath, C.B.; Burrell, L.M.; Angus, J.A. Liver disease and the renin-angiotensin system: Recent discoveries and clinical implications. J. Gastroenterol. Hepatol. 2008, 23, 1327–1338.
- Sansoè, G.; Aragno, M.; Mastrocola, R.; Mengozzi, G.; Novo, E.; Parola, M. Role of Chymase in the Development of Liver Cirrhosis and Its Complications: Experimental and Human Data. PLoS ONE 2016, 11, e0162644.
- Komeda, K.; Takai, S.; Jin, D.; Tashiro, K.; Hayashi, M.; Tanigawa, N.; Miyazaki, M. Chymase inhibition attenuates tetrachloride-induced liver fibrosis in hamsters. Hepatol. Res. 2010, 40, 832–840.
- Steckelings, U.M.; Larhed, M.; Hallberg, A.; E Widdop, R.; Jones, E.S.; Wallinder, C.; Namsolleck, P.; Dahlöf, B.; Unger, T. Non-peptide AT2-receptor agonists. Curr. Opin. Pharmacol. 2011, 11, 187–192.
- De Macêdo, S.M.; Guimarães, T.A.; Feltenberger, J.D.; Santos, S.H.S. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 2014, 62, 189–196.
- Prasad, A.; A Quyyumi, A. Renin-Angiotensin System and Angiotensin Receptor Blockers in the Metabolic Syndrome. Circulation 2004, 110, 1507–1512.
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circ. Res. 2000, 87, E1–E9.
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-converting Enzyme: Cloning and Functional Expression as a Captopril-Insensitive Carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243.
- Schiavone, M.T.; Santos, R.A.; Brosnihan, K.B.; Khosla, M.C.; Ferrario, C.M. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1–7) heptapeptide. Proc. Natl. Acad. Sci. USA 1988, 85, 4095–4098.
- Chappell, M.C. The Angiotensin-(1–7) Axis: Formation and Metabolism Pathways. Angiotensin-(1–7) 2019, 1–26.
- Santos, R.A.; e Silva, A.C.S.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263.
- Santos, R.A.; Haibara, A.S.; Campagnole-Santos, M.J.; Simões e Silva, A.C.; Paula, R.D.; Pinheiro, S.V.; de Fátima Leite, M.; Lemos, V.S.; Silva, D.M.; Guerra, M.T.; et al. Characterization of a New Selective Antagonist for Angiotensin-(1–7), d-Pro7-Angiotensin-(1–7). Hypertension 2003, 41, 737–743.
- Silva, D.; Vianna, H.; Cortes, S.; Campagnole-Santos, M.; Santos, R.; Lemos, V.S. Evidence for a new angiotensin-(1–7) receptor subtype in the aorta of Sprague–Dawley rats. Peptides 2007, 28, 702–707.
- Gembardt, F.; Grajewski, S.; Vahl, M.; Schultheiss, H.-P.; Walther, T. Angiotensin metabolites can stimulate receptors of the Mas-related genes family. Mol. Cell. Biochem. 2008, 319, 115–123.
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A.; et al. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111.
- Herath, C.B.; Mak, K.; Burrell, L.M.; Angus, P.W. Angiotensin-(1–7) reduces the perfusion pressure response to angiotensin II and methoxamine via an endothelial nitric oxide-mediated pathway in cirrhotic rat liver. Am. J. Physiol. Liver Physiol. 2013, 304, G99–G108.
- Tetzner, A.; Gebolys, K.; Meinert, C.; Klein, S.; Uhlich, A.; Trebicka, J.; Villacañas, Ó.; Walther, T. G-Protein–Coupled Receptor MrgD Is a Receptor for Angiotensin-(1–7) Involving Adenylyl Cyclase, cAMP, and Phosphokinase. Hypertension 2016, 68, 185.
- Bader, M. Tissue Renin-Angiotensin-Aldosterone Systems: Targets for Pharmacological Therapy. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 439–465.
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14.
- Bataller, R.; Sancho-Bru, P.; Gines, P.; Lora, J.M.; Al-Garawi, A.; Solé, M.; Colmenero, J.; Nicolás, J.M.; Jiménez, W.; Weich, N.; et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 2003, 125, 117–125.
- Lang, C.C.; Struthers, A.D. Targeting the renin-angiotensin-aldosterone system in heart failure. Nat. Rev. Cardiol. 2013, 10, 125–134.
- Nistala, R.; Wei, Y.; Sowers, J.R.; Whaley-Connell, A. Renin-angiotensin-aldosterone system-mediated redox effects in chronic kidney disease. Transl. Res. 2009, 153, 102–113.
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 2003, 112, 1383–1394.
- Lubel, J.S.; Herath, C.B.; Tchongue, J.; Grace, J.; Jia, Z.; Spencer, K.; Casley, D.; Crowley, P.; Sievert, W.; Burrell, L.M.; et al. Angiotensin-(1–7), an alternative metabolite of the renin–angiotensin system, is up-regulated in human liver disease and has antifibrotic activity in the bile-duct-ligated rat. Clin. Sci. 2009, 117, 375–386.
- Paizis, G.; Cooper, M.E.; Schembri, J.M.; Tikellis, C.; Burrell, L.M.; Angus, P.W. Up-regulation of components of the renin-angiotensin system in the bile duct–ligated rat liver. Gastroenterology 2002, 123, 1667–1676.
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of Local Renin-Angiotensin Systems. Physiol. Rev. 2006, 86, 747–803.
- Herath, C.B.; Warner, F.J.; Lubel, J.S.; Dean, R.G.; Jia, Z.; Lew, R.A.; Smith, A.I.; Burrell, L.M.; Angus, J.A. Upregulation of hepatic angiotensin-converting enzyme 2 (ACE2) and angiotensin-(1–7) levels in experimental biliary fibrosis. J. Hepatol. 2007, 47, 387–395.
- Mak, K.Y.; Chin, R.; Cunningham, S.C.; Habib, M.R.; Torresi, J.; Sharland, A.F.; E Alexander, I.; Angus, J.A.; Herath, C.B. ACE2 Therapy Using Adeno-associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol. Ther. 2015, 23, 1434–1443.
- Rajapaksha, I.G.; Mak, K.Y.; Huang, P.; Burrell, L.M.; Angus, J.A.; Herath, C.B. The small molecule drug diminazene aceturate inhibits liver injury and biliary fibrosis in mice. Sci. Rep. 2018, 8, 1–14.
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172.
- Bataller, R.; Gäbele, E.; Parsons, C.J.; Morris, T.; Yang, L.; Schoonhoven, R.; A Brenner, D.; Rippe, R.A. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology 2005, 41, 1046–1055.
- Herath, C.B.; Mak, K.Y.; Angus, P.W. Role of the Alternate RAS in Liver Disease and the GI Tract. In The Protective Arm of the Renin Angiotensin System (RAS); Elsevier: Amsterdam, The Netherlands, 2015; pp. 239–247.
- Bataller, R.; Ginès, P.; Nicolás, J.M.; Görbig, M.; Garcia–Ramallo, E.; Gasull, X.; Bosch, J.; Arroyo, V.; Rodés, J. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 2000, 118, 1149–1156.
- Deleve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746.
- Maretti-Mira, A.C.; Wang, X.; Wang, L.; DeLeve, L.D. Role of incomplete stem cell maturation in hepatic fibrosis. In HEPATOLOGY; Wiley-Blackwell: Hoboken, NJ, USA, 2016.
- Jarnagin, W.R.; Rockey, D.C.; E Koteliansky, V.; Wang, S.S.; Bissell, D.M. Expression of variant fibronectins in wound healing: Cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J. Cell Biol. 1994, 127, 2037–2048.
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617.
- Jonsson, J.R.; Clouston, A.D.; Ando, Y.; Kelemen, L.I.; Horn, M.J.; Adamson, M.D.; Purdie, D.M.; Powell, E.E. Angiotensin-Converting Enzyme Inhibition Attenuates the Progression of Rat Hepatic Fibrosis. Gastroenterology 2001, 121, 148–155.
- Turkay, C.; Yönem, Ö.; Arıcı, S.; Koyuncu, A.; Kanbay, M.; Arici, S. Effect of Angiotensin-converting Enzyme Inhibition on Experimental Hepatic Fibrogenesis. Dig. Dis. Sci. 2007, 53, 789–793.
- Paizis, G.; Gilbert, R.E.; Cooper, M.E.; Murthi, P.; Schembri, J.M.; Wu, L.L.; Rumble, J.R.; Kelly, D.J.; Tikellis, C.; Cox, A.; et al. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J. Hepatol. 2001, 35, 376–385.
- Yi, E.-T.; Liu, R.-X.; Wen, Y.; Yin, C.-H. Telmisartan attenuates hepatic fibrosis in bile duct-ligated rats. Acta Pharmacol. Sin. 2012, 33, 1518–1524.
- Wei, H.-S.; Li, D.-G.; Lu, H.-M.; Zhan, Y.-T.; Wang, Z.-R.; Huang, X.; Zhang, J.; Cheng, J.-L.; Xu, Q.-F. Effects of AT1 receptor antagonist, losartan, on rat hepatic fibrosis induced by CCl4. World J. Gastroenterol. 2000, 6, 540–545.
- Hirose, A.; Ono, M.; Saibara, T.; Nozaki, Y.; Masuda, K.; Yoshioka, A.; Takahashi, M.; Akisawa, N.; Iwasaki, S.; Oben, J.A.; et al. Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology 2007, 45, 1375–1381.
- Corey, K.E.; Shah, N.; Misdraji, J.; Abu Dayyeh, B.K.; Zheng, H.; Bhan, A.K.; Chung, R.T. The effect of angiotensin-blocking agents on liver fibrosis in patients with hepatitis C. Liver Int. 2009, 29, 748–753.
- Stokkeland, K.; Lageborn, C.T.; Höijer, J.; Bottai, M.; Stål, P.; Söderberg-Löfdal, K.; Ekbom, A. Statins and Angiotensin-Converting Enzyme Inhibitors are Associated with Reduced Mortality and Morbidity in Chronic Liver Disease. Basic Clin. Pharmacol. Toxicol. 2018, 122, 104–110.
- Kim, M.Y.; Cho, M.Y.; Baik, S.K.; Jeong, P.H.; Suk, K.T.; Jang, Y.O.; Yea, C.J.; Kim, J.W.; Kim, H.S.; Kwon, S.O.; et al. Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis—A randomized open-label controlled study. Liver Int. 2012, 32, 977–987.
- Hidaka, H.; Nakazawa, T.; Shibuya, A.; Minamino, T.; Takada, J.; Tanaka, Y.; Okuwaki, Y.; Watanabe, M.; Koizumi, W. Effects of 1-year administration of olmesartan on portal pressure and TGF-beta1 in selected patients with cirrhosis: A randomized controlled trial. J. Gastroenterol. 2011, 46, 1316–1323.
- Debernardi-Venon, W.; Martini, S.; Biasi, F.; Vizio, B.; Termine, A.; Poli, G.; Brunello, F.; Alessandria, C.; Bonardi, R.; Saracco, G.; et al. AT1 receptor antagonist Candesartan in selected cirrhotic patients: Effect on portal pressure and liver fibrosis markers. J. Hepatol. 2007, 46, 1026–1033.
- Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Kaji, K.; Douhara, A.; Yamao, J.; Toyohara, M.; Mitoro, A.; Sawai, M.; Yoshida, M.; et al. Combination of branched-chain amino acid and angiotensin-converting enzyme inhibitor improves liver fibrosis progression in patients with cirrhosis. Mol. Med. Rep. 2011, 5, 539–544.
- Terui, Y.; Saito, T.; Watanabe, H.; Togashi, H.; Kawata, S.; Kamada, Y.; Sakuta, S. Effect of angiotensin receptor antagonist on liver fibrosis in early stages of chronic hepatitis C. Hepatology 2002, 36, 1022.
- McPherson, S.; Wilkinson, N.; Tiniakos, D.; Wilkinson, J.; Burt, A.D.; McColl, E.; Stocken, D.D.; Steen, N.; Barnes, J.; Goudie, N.; et al. A randomised controlled trial of losartan as an anti-fibrotic agent in non-alcoholic steatohepatitis. PLoS ONE 2017, 12, e0175717.
- Zhu, Q.; Li, N.; Li, F.; Zhou, Z.; Han, Q.; Lv, Y.; Sang, J.; Liu, Z. Therapeutic effect of renin angiotensin system inhibitors on liver fibrosis. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17.
- Runyon, B.A. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology 2013, 57, 1651–1653.
- EASL. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460.

