 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Samo Ribarič | + 1803 word(s) | 1803 | 2021-01-28 05:07:55 | | | |
| 2 | Dean Liu | Meta information modification | 1803 | 2021-03-03 02:38:41 | | |
Video Upload Options
Nanotechnology (NT) enables a new, alternative pathway for development of AD treatment interventions in human. At present, the NT treatments for attenuation of AD memory impairment are at the animal model stage. They are faced with the twin challenges of the nature of AD: a chronic impairment unique to human and the incomplete understanding of AD′s aetiology.
1. Introduction
Nanotechnology (NT) provides a new approach to develop alternative drug delivery treatments for all stages of Alzheimer’s disease (AD). NT uses particles with at least one dimension smaller than 100 nm, the nano particles (NPs)[1]. The International Organization for Standardization defines NPs as nano-objects with all three external dimensions in the nanoscale[2].
The NPs have several advantages compared to traditional drug delivery compounds. They have a very small size with a high surface-to-volume ratio that facilitates interactions with biomolecules. They can be produced to different shapes (spherical, cubic, rod-like) and sizes to modify their movement across biological barriers. NPs can be used either for disease diagnosis or for treatment. They can bind with a wide variety of desired ligands (by adsorbing, entrapping or covalent bonding) to acquire new diagnostic, therapeutic or physiological properties, including the ability to cross the blood-brain barrier (BBB)[1].
NPs, for AD treatment or diagnosis, are either natural-polymer based, synthetic-polymer based or inorganic substances. Examples of synthetic-polymer based NPs are poly (ethylenimine), poly-(alkylcyanoacrylates), poly (amidoamine) dendrimers, poly (ε-caprolactone), poly (D, L-lactic acid) (PLA), poly (lactic-co-glycolic acid) (PLGA), polyethylene glycol (PEG), and polyesters (poly (lactic acid) (PLA)). Examples of inorganic materials for therapeutic NPs are gold, silica, carbon. The desired size and shape of NPs is more difficult to achieve from polymeric than from inorganic materials. A faster degradation and elimination from the body through the kidneys, and a lower risk of toxicity make polymeric NPs more suitable for human therapeutic applications than inorganic NPs. Natural-polymer based NPs, such as amino acids (poly(lysine), poly (aspartic acid), polysaccharides (chitosan and alginate) and proteins (gelatine and albumin), have the ability to interact with specific receptors/transporters expressed by endothelial cells combined with the disadvantages of poor structure modification and tracking by imaging platforms. Inorganic NPs are most suitable for imaging applications, due to their long half-life and inherent chemical stability in the biological environment. The desired physio-chemical properties of NPs, for optimal therapeutic efficiency, are: a size between 50 and 100 nm, a spherical shape, a close to zero (low positive charge) or negative zeta potential[3].
NPs tend to adsorb proteins in body fluids and form a protein coating. For example, gold NPs adsorb more than 70 different serum proteins. This protein layer modulates the NPs′ ability to interact with the environment, changes the NPs′ physicochemical properties, aggregation rate, half-life and in case of blood borne NPs, enhances their sequestration in spleen and liver. Coating is attenuated by grafting NPs with PEG and also increases NPs′ blood half-life[3].
The BBB controls bidirectional transport of biomolecules between blood vessels and brain cells. Crossing the BBB is a significant challenge for the development of drug delivery systems to the brain. The physicochemical properties of NP are modified, by attaching different ligands, with optimal ligand density and receptor affinity, to their surface to facilitate drug delivery. Examples of ligands that facilitate BBB penetration are: (a) Ligands that interact directly with BBB receptors or transporters-poly(sorbate 80, alias, Tween 80) with adsorbed apolipoprotein E and/or A-I from the blood stream; (b) ligands with direct interaction with BBB receptors or transporters-for transferrin or insulin receptor, or glucose transporter; (c) ligands that increase the NPs′ charge and hydrophobicity-amphiphilic peptides facilitating uptake by BBB endothelial cells; and (d) ligands that improve blood circulation time-for example, PEG or PEG–PLGA [1][2][3][4][5][6][7][8][9]. Classification and properties of NPs, evaluated for AD treatment and diagnostic interventions, are presented in Table 1[8][10][11][12][13].
Table 1. Classification and properties of nanoparticles tested for AD treatment or diagnosis.
| Nanoparticle Types |
Core Structures | Surface Modifications | Cargo |
|---|---|---|---|
| 3-dimensional DNA nanostructures | tetrahedral DNA nanostructures | ||
| Carbon nanotubes | single-walled carbon nanotubes; multi-walled carbon nanotubes |
anti-Tau antibody; gold & antibody-binding protein & Aβ antibody; polysorbate or phospholipid coating |
acetylcholine; berberine |
| Carbon quantum dots | polymerised o-phenylenediamine quasispheroidal carbon based nanomaterial of quantum size; | ||
| Dendrimers | gallic acid-triethylene glycol; cationic phosphorous dendrimers;poly-amidoamine; poly-propylene-imine |
helical β-peptide foldamers; maltose; morpholine groups; tetra-maleimidopropionyl |
|
| Gold | gold nanoparticles; gold nanorods |
carboxyl groups conjugated to nanoparticles; N-terminal cysteine peptide conjugated to gold nanorods; |
|
| Lipid nanoparticles | solid lipid nanoparticle; nanostructured lipid carrier |
monoclonal antibodies to transferrin receptors on BBB; pluronic acid; polyethylene glycol and lactoferrin; polysorbate 80 |
BACE1 siRNA; curcumin; donepezil; galantamine; resveratrol |
| Liposomes | cholesterol and phosphatidyl-choline; cholesterol and 1,2-distearoyl-sn-glycero-3-phosphocholine; cholesterol and sphingomyelin |
cell penetration peptides and polyethylene glycol; phosphatidic acid and Apo-E; phosphatidic acid; polyethylene glycol |
curcumin; galantamine; rivastigmine |
| Magnetic nanoparticles | gadopentetic acid; iron oxide (Fe3O4; Fe3O3); magnetite/ceria nanoparticles; polysiloxane matrix with gadolinium chelates; |
chitosan and IgG-anti-amyloid antibodies; curcumin and polyethylene glycol and polyvinylpyrrolidone; Aβ oligomers monoclonal antibodies and polyethylene glycol and nitro-L-DOPA; Aβ-antibodies and polyethylene glycol |
cyclophos-phamide |
| Polymeric nanoparticles | Poly (lactic-co-glycolic acid); Poly (lactic acid); chitosan; amino-group-modified mesoporous silica nanoparticles; selenium-(poly-lactide-co-glycolide) nanospheres |
polyethylene glycol; polysorbate 80; polyethylene glycol and Aβ-binding peptide and targeting peptide to overcome blood brain barrier; tau-binding peptide and iron oxide and ceria nanocrystals |
curcumin; galantamine; rivastigmine; tacrine; methylene blue |
| Silver | silver nanoparticles | / | / |
| Sulphur | volute, tadpole or sphere-like nanoparticles | / | / |
The main transport pathways for NPs across the BBB are receptor mediated transcytosis and adsorptive mediated transcytosis[1][14] NPs can cross the BBB by several pathways. For example, liposomes can cross the BBB by either receptor mediated transcytosis or adsorptive mediated transcytosis[7]. Selected types of potential NP BBB pathways are presented in Figure 1[1][5][6][7][15][16].
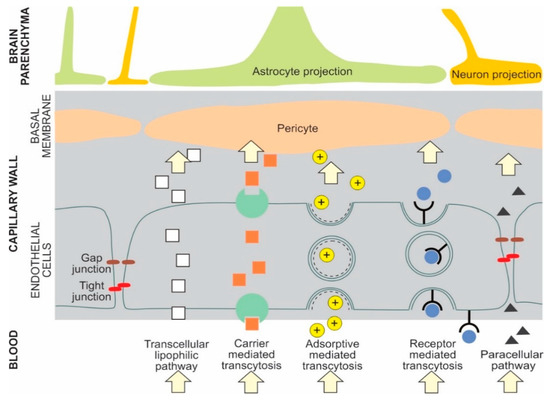
Figure 1. Examples of potential NP transport pathways across the blood-brain barrier.
NPs are increasingly recognized as promising candidates for new AD therapies[3][4][6][17]. The amyloid cascade hypothesis still underpins the development of Aβ-related diagnosis/treatment approaches. However, research has shifted to a multifactorial aetiology approach to AD, recognising the unique temporal contributions of (a) Aβ1-42 accumulation, formation and accumulation of toxic, soluble Aβ oligomers (AβOs); (b) the binding of zinc, copper, and iron cations to Aβ1-42 peptides that accelerates formation of AβOs; (c) tau protein phosphorylation, and (d) oxidative stress and chronic neuroinflammation elicited and sustained by glial cells. The future of NP-based treatments of AD is in developing treatment interventions tailored to each of the four AD stages: (a) The asymptomatic, preclinical stage; (b) the progressively symptomatic mild cognitive impairment (MCI); (c) the mild to moderate dementia; and (d) the severe dementia[3][4][6][8].
The mechanistic approaches to development of AD therapies, with NPs carrying therapeutic agents, are: (a) the clearance of Aβ fibrils/aggregates; (b) the development of acetylcholinesterase inhibitors loaded NPs to ameliorate cholinergic system impairment; (c) the attenuation of neuroinflammation; (d) the attenuation of tau hyperphosphorylation; (e) the development of anti-Aβ peptide antibodies loaded on circulating NPs that initiate ‘the sink mechanism’, by removing the soluble Aβ peptides from the brain to the blood circulation.
2. Alzheimer′s Disease and Memory Impairment
2.1. Short-Term and Long-Term Memory
Memory formation and retrieval are essential brain functions supporting human′s daily activities. The key process enabling memory retention is the conversion of short-term memory (STM) to long-term memory (LTM). STM and LTM are formed and supported by distinct neurobiological processes. STM is underpinned by modulated activity patterns of existing brain neural networks and their post-translational modifications of proteins (e.g., protein phosphorylation). LTM is underpinned by structural and functional changes of neural networks elicited by new gene expression (e.g., an increase of the number and size of synaptic connections among specific brain neural networks)[17][18].
2.2. Memory Impairment
Memory impairment, including the degraded formation and recall of memories, can be present in the acute or chronic brain disorders. In human brain disorders, up to four distinct memory modalities can be affected: Episodic, semantic, working, and procedural memory —each with specific clinical signs of memory loss, affected neuroanatomical networks, and commonly associated acute or chronic disorders. Consciously recalled memories of events, objects or facts are labelled as declarative memories, and their formation is critically dependent on normal function of hippocampus and medial temporal lobes[17]. Examples of declarative memories are: Episodic, semantic, and working memories. Episodic memory impairment, the inability to recall recent personal events, is one of the most often perceived forms of memory dysfunction in human. This memory impairment is usually associated with hippocampus and medial temporal lobes dysfunction; however, other brain structures also participate in episodic memory formation: diencephalon, limbic system, posterior cingulate and precuneus region[19]. Progressive memory impairment, ultimately contributing to dementia, that is associated with pathological changes of hippocampus, medial temporal lobes, posterior cingulate or precuneus region, is a hallmark of Alzheimer’s disease[20][21][22].
2.3. Alzheimer′s Disease, Dementia and Memory Impairment
The most prevalent symptom of AD in humans is the cognitive impairment[23]. Also, AD is the most common cause of human dementia, a state of severe cognitive impairment affecting memory, thinking, and behaviour that prevents the patient to independently perform everyday activities. The projected number of AD patients will triple between 2013 and 2023[24]. Two sub-groups of AD are identified. The early-onset form, clinically diagnosed before 65 years of age, a polygenic form where only 10% of the early-onset cases are attributable to the altered gene expression of either amyloid precursor protein, presenilin-1 or presenilin-2, affecting about 1% of all AD patients. The best understood early-onset forms of AD are the familial early-onset forms (efAD) with mutations in expression of amyloid precursor protein, presenilin-1 or presenilin-2. The late-onset, sporadic form (sAD), is clinically diagnosed before 65 years of age and affects 99% of all AD patients[25]. The apolipoprotein E4 (Apo-E4) gene is a known risk factor for the late-onset AD, increasing the risk by up to 10-fold[26]. Individuals with Apo-E2 or Apo-E3 gene have a higher synaptic plasticity and repair capacity compared to the non Apo-E2, non Apo-E3 population[27]. The AD diagnosis is unequivocally confirmed only post-mortem by the brain atrophy associated with; (a) extracellular senile plaques composed of Aβ peptides in various stages of aggregation (i.e., amyloid deposits) and (b) intraneuronal neurofibrillary tangles (NFTs), composed of hyperphosphorylated tau protein. In human, these characteristic intracellular and extracellular lesions first appear in the hippocampus and entorhinal cortex (the main interface between hippocampus and neocortex). The entorhinal cortex–hippocampus system underpins episodic memories, especially the formation, consolidation, and sleep optimisation of spatial memories. Later, the AD associated lesions spread to include the temporal, parietal, and frontal association cortices[28]. The AD memory impairment is assumed to occur when the progressive reduction in brain synaptic density abolishes the cognitive reserve (CR). The CR varies in size, from person to person, and explains the variability in memory decline among AD patients with similar brain pathology. Paradoxically, a later appearance in clinical signs of dementia, due to a high CR, is followed by a faster progression of memory decline[29][30].
References
- Athira, S.; Nadukkandy, P.; Mohanan, P. Interaction of nanoparticles with central nervous system and its consequences. Am. J. Res. Med. Sci. 2018, 4, 12.
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Volceanov, A.; Teleanu, R.I. Impact of nanoparticles on brain health: An up to date overview. J. Clin. Med. 2018, 7, 490.
- Saraiva, C.; Praca, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47.
- Hajipour, M.J.; Santoso, M.R.; Rezaee, F.; Aghaverdi, H.; Mahmoudi, M.; Perry, G. Advances in alzheimer’s diagnosis and therapy: The implications of nanotechnology. Trends Biotechnol. 2017, 35, 937–953.
- Pires, P.C.; Santos, A.O. Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies. J. Control. Release 2018, 270, 89–100.
- Gupta, J.; Fatima, M.T.; Islam, Z.; Khan, R.H.; Uversky, V.N.; Salahuddin, P. Nanoparticle formulations in the diagnosis and therapy of alzheimer’s disease. Int. J. Biol. Macromol. 2019, 130, 515–526.
- Fonseca-Santos, B.; Gremiao, M.P.; Chorilli, M. Nanotechnology-based drug delivery systems for the treatment of alzheimer’s disease. Int. J. Nanomed. 2015, 10, 4981–5003.
- de la Torre, C.; Cena, V. The delivery challenge in neurodegenerative disorders: The nanoparticles role in alzheimer’s disease therapeutics and diagnostics. Pharmaceutics 2018, 10, 190.
- Wong, K.H.; Riaz, M.K.; Xie, Y.N.; Zhang, X.; Liu, Q.; Chen, H.J.; Bian, Z.X.; Chen, X.Y.; Lu, A.P.; Yang, Z.J. Review of current strategies for delivering alzheimer’s disease drugs across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 381.
- Greish, K.; Alqahtani, A.A.; Alotaibi, A.F.; Abdulla, A.M.; Bukelly, A.T.; Alsobyani, F.M.; Alharbi, G.H.; Alkiyumi, I.S.; Aldawish, M.M.; Alshahrani, T.F.; et al. The effect of silver nanoparticles on learning, memory and social interaction in balb/c mice. Int. J. Env. Res. Public Health 2019, 16, 148.
- Sun, J.; Xie, W.; Zhu, X.; Xu, M.; Liu, J. Sulfur nanoparticles with novel morphologies coupled with brain-targeting peptides rvg as a new type of inhibitor against metal-induced abeta aggregation. ACS Chem. Neurosci. 2018, 9, 749–761.
- Kim, D.; Kwon, H.J.; Hyeon, T. Magnetite/ceria nanoparticle assemblies for extracorporeal cleansing of amyloid-beta in alzheimer’s disease. Adv. Mater. 2019, 31, e1807965.
- Chen, Q.; Du, Y.; Zhang, K.; Liang, Z.; Li, J.; Yu, H.; Ren, R.; Feng, J.; Jin, Z.; Li, F.; et al. Tau-targeted multifunctional nanocomposite for combinational therapy of alzheimer’s disease. ACS Nano 2018, 12, 1321–1338.
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 20.
- Pulgar, V.M. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front. Neurosci. 2018, 12, 1019.
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert. Opin. Drug. Deliv. 2019, 16, 271–285.
- Bisaz, R.; Travaglia, A.; Alberini, C.M. The neurobiological bases of memory formation: From physiological conditions to psychopathology. Psychopathology 2014, 47, 347–356.
- Mayford, M.; Siegelbaum, S.A.; Kandel, E.R. Synapses and memory storage. Cold. Spring. Harb. Perspect. Biol. 2012, 4, a005751.
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The physiological roles of amyloid-beta peptide hint at new ways to treat alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 118.
- Matthews, B.R. Memory dysfunction. Continuum 2015, 21, 613–626.
- Sperling, R.A.; Dickerson, B.C.; Pihlajamaki, M.; Vannini, P.; LaViolette, P.S.; Vitolo, O.V.; Hedden, T.; Becker, J.A.; Rentz, D.M.; Selkoe, D.J.; et al. Functional alterations in memory networks in early alzheimer’s disease. Neuromol. Med. 2010, 12, 27–43.
- de Ipolyi, A.R.; Rankin, K.P.; Mucke, L.; Miller, B.L.; Gorno-Tempini, M.L. Spatial cognition and the human navigation network in ad and mci. Neurology 2007, 69, 986–997.
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175.
- Wimo, A.; Jonsson, L.; Winblad, B. An estimate of the worldwide prevalence and direct costs of dementia in 2003. Dement. Geriatr. Cogn. Disord. 2006, 21, 175–181.
- Wingo, T.S.; Cutler, D.J.; Wingo, A.P.; Le, N.A.; Rabinovici, G.D.; Miller, B.L.; Lah, J.J.; Levey, A.I. Association of early-onset alzheimer disease with elevated low-density lipoprotein cholesterol levels and rare genetic coding variants of apob. JAMA Neurol. 2019, 76, 809–817.
- Mayeux, R. Gene-environment interaction in late-onset alzheimer disease: The role of apolipoprotein-epsilon4. Alzheimer Dis. Assoc. Disord. 1998, 12 (Suppl. 3), S10–S15. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9876937 (accessed on 21 January 2021).
- Bufill, E.; Carbonell, E. Apolipoprotein e polymorphism and neuronal plasticity. Am. J. Hum. Biol. 2006, 18, 556–558.
- Holger, J. Memory loss in alzheimer’s disease. Clin. Res. 2013, 15, 445–454.
- Terry, R.D.; Katzman, R. Life span and synapses: Will there be a primary senile dementia? Neurobiol. Aging 2001, 22, 347–348.
- van Loenhoud, A.C.; Wink, A.M.; Groot, C.; Verfaillie, S.C.J.; Twisk, J.; Barkhof, F.; van Berckel, B.; Scheltens, P.; van der Flier, W.M.; Ossenkoppele, R. A neuroimaging approach to capture cognitive reserve: Application to alzheimer’s disease. Hum. Brain Mapp. 2017, 38, 4703–4715.

