 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Barbara Molon | + 3218 word(s) | 3218 | 2021-02-20 04:01:52 | | | |
| 2 | Camila Xu | Meta information modification | 3218 | 2021-02-25 10:15:20 | | |
Video Upload Options
TGF-β is a well know cytokine and growth factor related to tumor growth and fibrosis in different kind of cancers such as lung, pancreas, colon cancer and hepatocellular carcinoma.
1. Transforming Growth Factor Beta Signaling in Tumors
TGF-β is a well know cytokine and growth factor related to tumor growth and fibrosis in different kind of cancers such as lung, pancreas, colon cancer and hepatocellular carcinoma [9]. Only recently, its effect on the tumor and TME metabolic reprograming has been revealed. TGF-β is a member of a protein family that includes activin, bone morphogenic proteins (BMP) and Nodal [10] and it is expressed in three different isoforms (TGF-β1, TGF-β2 and TGF-β3). TGF-β is synthetized as inactive form composed by a C-terminal domain and the latency-associated peptide (LAP) at the N-terminal. The circulating TGF-β binds through the LAP portion the latent TGF-β binding proteins (LTBPs) leading to the assembling of the large latent complex (LLC) [11]. The release of the active TGF-β from the LCC is dependent on the action of cell-surface transmembrane receptor proteins such as αvβ6 and αvβ8 integrins. Indeed, once bounded to the RDG sequence in the LAP, these proteins unfold the complex and induce the release of the active TGF-β [12]. Extracellular matrix (ECM) proteins involved in this process further include Thrombospondin 1 [13], Glycoprotein A repetitions predominant protein (GARP) [14], Leucine-rich repeat containing protein 33 (LRRC33) [15], Cathepsin D and metalloproteases (MMPs) such as MMP-9 and MMP14 [16]. Once released, active TGF-β directly associates to its heteromeric serine/threonine kinase receptor (TβRI-TβRII) [17], often assisted by accessory coreceptors as TβRIII or β-glycan [18]. Then, the TGF-β induced TβRII autophosphorylation, at the Ser213 and Ser409, determines its kinase activity on TβRI GS domain triggering two distinct downstream pathways, called canonical and non-canonical pathways [19]. In the SMAD-dependent canonical pathway, TβRI phosphorylates SMAD2 and SMAD3 to form an heteromeric complex with SMAD4. This complex accumulates into the nucleus where the expression of several genes is controlled by directly binding several DNA regulatory regions [20]. On the other side, the TGF-β non-canonical pathway activates GRB2, SOS or TAK1 leading to the MAPK kinases cascade that, in turn, regulates NF-κB, c-FOS, c-JUN c-MYC transcription factors [21]. Beside the transcriptional regulation, TGF-β-triggered circuits directly induce cytoskeleton remodeling by activation of small protein GTP-ases proteins such as RhoA, Rock or by Cofilin-1 signaling [22]. The duality of TGF-β signaling has been repeatedly documented in cancer as this cytokine acts as a potent tumor suppressor during early tumor development, but paradoxically, it become a metastatic promoter in later stages [23]. As extensively reviewed elsewhere [23], several studies have been developed to specifically manage this challenging target for cancer treatment [23].
Among many other documented effects, the canonical TGF-β/SMAD pathway has been linked to the epithelial-mesenchymal transition (EMT), that contribute to the metastatic spreading in not all, but many tumors. In pre-clinical breast cancer models the activation of SNAIL1-SMAD3/4 transcriptional complex regulated gene expression during EMT [24]. Further evidence showed that TGF-β–induced activation of mTOR and the PI3K–Akt pathway correlated with increased cell size and protein content in NMuMG mammary cells, supporting an important role for non-Smad TGF-β signaling pathway in cancer progression [25]. In this regard, a recent report indicated that a prolonged TGF-β exposure promoted stable EMT in mammary epithelial and carcinoma cells, by triggering the mTOR pathway; this stable EMT phenotype contributed to controlling cancer cell stemness drug resistance of breast cancer cells [26]. TGF-β cascades can differently act on multiple cell compartments within the bulk tumor, mainly establishing a pro-tumoral metabolic reprogramming of the TME components. In the TME, TGF-β can be produced by several cell types including tumor, stromal and immune cells mediating both autocrine and paracrine effects. Moreover, distant TGF-β sites of release can not be excluded in several cancers. Indeed, in most of the in vivo studies, technical impairments limited the precise definition of TGF-β release origin. In this manuscript, we will focus on the TGF-β impact on cancer-associated fibroblasts (CAFs), endothelial and immune cells, focusing on metabolic pathways (Figure 1).
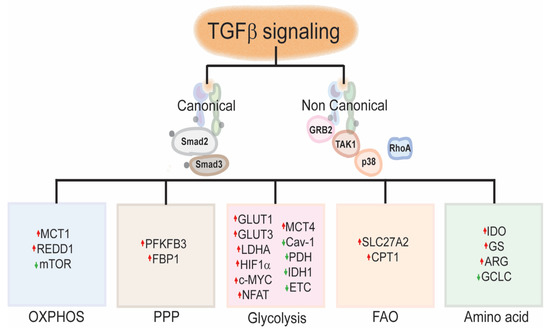
Figure 1. TGF-β signaling and metabolic reprograming: TGF-β canonical and non-canonical signaling activation promotes the metabolic skewing to either glycolytic or oxidative metabolism in the TME by modulating the transcription of genes involved in glycolysis, OXPHOS, PPP, FAO or amino acid metabolism, directly or indirectly (see text.) TGFβ, Transforming Growth factor beta; GRB2, Growth factor receptor-bound protein 2; TAK1, Mitogen-activated protein kinase kinase kinase 7; p38, p38 mitogen-activated protein kinases; RhoA, Ras homolog family member A; MCT1, Monocarboxylate transporter 1; MCT4, Monocarboxylate transporter 4; REDD1, Regulated in development and DNA damage response 1; mTOR, mechanistic target of rapamycin; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; FBP1, Fructose-1,6-biphosphatase, GLUT1, Glucose transporter 1; GLUT3, Glucose transporter 3; LDHA, Lactate dehydrogenase A; HIF1α, Hypoxia-inducible factor 1 alpha; c-MYC, MYC proto-oncogene BHLH Transcription factor; NFAT; Nuclear factor of activated T-cells; Cav-1, Caveolin 1; PDH Pyruvate dehydrogenase; IDH1, Isocitrate dehydrogenase 1; ETC, Electron transport chain; SLC27A2, solute carrier family 27 (fatty acid transporter), member 2; IDO, Indoleamine 2,3-dioxygenase; GS; Glutamine synthetase; ARG, Arginase; GCLC, Glutamate-cysteine Ligase catalytic subunit; OXPHOS, Oxidative phosphorylation; PPP, Pentose Phosphate Pathway; FAO, Fatty acid oxidation.
2. TGF-β Induced Cancer-Associated Fibroblast (CAF) Metabolic Reprogramming
2.1. CAF General Function
CAFs are heterogenous cells of the TME. Their differences mainly depend on multiple origins, various tissue localizations and distinct activation stages [27]. Despite the precise CAF origin is still unsolved, collected evidence defined that CAFs might derived from: (i) activated resident fibroblasts, (ii) engrafted and differentiated mesenchymal stromal cells and (iii) accumulated mutations in epithelial or tumor cells [28]. This heterogeneity troubles the definition of specific CAF markers. Consequently, the precise CAF tissue localization is not fully described [29]. Furthermore, overwhelming data pointed out a sub-specialization of CAFs possibly reflecting distinct functional states [30]. Nonetheless, CAFs are usually identified by the expression of α-smooth muscle actin (α-SMA), fibroblast specific protein, fibroblast activation protein (FAP) and platelet-derived growth factor receptors α and β (PDGF-α and PDGF-β) [29][31]. These cells have displayed multiple pro-tumorigenic functions cooperating with other CAF-independent mechanisms [32]. In particular, CAFs exhibit either a matrix-producing phenotype or an immunomodulating secretome, with a biunivocal functional connection of these effects [30]. CAFs release ECM proteins (such as type I, type III, type IV and type V collagens, many different laminins and fibronectin) as well as extracellular enzymes (MMPs and lysyl oxidase) representing the main contributors of the ECM remodeling [33]. This CAF-sculpted tumor ECM composition and organization leads to increased matrix stiffness responsible for higher cancer cell adhesion to ECM, enhanced cancer cell proliferation and invasion, and elevated interstitial fluid pressure promoting pro-angiogenic signatures [34][35][36]. Concomitantly, CAFs shape the immune microenvironment toward a tolerant and immunosuppressive milieu contributing to cancer immune escape [37]. This process is dependent on the CAF production of both immunosuppressive cytokines and immune checkpoint ligands. Thus, CAFs block the cytotoxic CD8+ T cell recruitment, and prompt tumor infiltrating inflammatory cell polarization toward an anti-inflammatory phenotype [37]. Together, CAF-remodeled matrix and CAF-established immunosuppression within the tumor enhance cancer invasiveness, stemness and proliferative behavior, favoring EMT and sustaining tumor anchorage and metastaticity [27]. Besides these processes, CAFs further support the insurgence of a pro-tumoral microenvironment by engaging a metabolic interplay within the TME. Here, TGF-β has a central reprogramming role in both the (i) CAF generation and (ii) the delineation of a pro-tumoral TME.
2.2. How Does TGF-β Directly Alter CAF Functions?
A synergistic interplay between neoplastic cell transformation and the surrounding environment is required for detrimental cancer progression [38]. CAF activation is pivotal during the TME definition, and TGF-β family ligands play a crucial role in this process. The critical contribution of TGF-β to CAF generation is dependent on a metabolic reprogram, that mainly attends to an unbalance in the oxidative framework [30]. Indeed, the TGF-β canonical and non-canonical pathways increase reactive oxygen species (ROS) production in CAFs by the impairment of the respiratory transport chain, specifically acting on Complex IV [39] by the GS3K action [40]. Furthermore, Smad signaling contributes to increase the expression of NOX4 [41], thus sustaining ROS accumulation. Additionally, TGF-β-mediated transcriptional competition for several DNA-binding sites, as Glutamate-Cysteine Ligase (GSL) gene, determines glutathione depletion [42] and antioxidant NRF2 impairment [43], eventually contributing to the oxidative stress. ROS increase in fibroblasts modulates CAF markers as α-SMA expression [44] and it is linked to the loss of stromal caveolin-1 (Cav-1), due to an extreme induction of LC3B-II-mediated lysosomal degradation [45]. Beside the ROS-dependent mechanism, TGF-β activation increases the intracellular accumulation of α-ketoglutarate by downregulating the isocitrate dehydrogenase 1 (IDH1). This event further contributes to Cav-1 downregulation [46]. Notably, the loss of stromal Cav-1 within the tumor fuels cancer progression through various paracrine signaling mechanisms, including the initiation of CAF-mediated ECM remodeling [47]. Moreover, it has been reported that the attenuation of Cav-1 pushes fibroblasts toward an autophagic phenotype with elevate aerobic glycolysis. Indeed, the down expression of Cav-1 contributes to the mitochondrial disfunction thus creating a positive-loop between Cav-1 and ROS production [48]. Reduced expression of Cav-1 also triggers the down regulation and activity of prolyl hydroxylase domain-containing protein (PHD) [49], which is involved in the hydroxylation of HIF1α and IκB. The consequent HIF stabilization and NFκB activation leads to glycolytic gene transcription such as lactate dehydrogenase A (LDHA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [50]. All together, these steps determine the CAF secretion of high energy metabolites such as pyruvate and lactate compelling mitochondrial OXPHOS in cancer cells [51]. Along with Cav-1/glycolysis circuit, emerging data pointed out the TGF-β dependent autophagy-to-senescence transition (AST) of some TME component, especially CAFs [52]. In this scenario, autophagic fibroblasts enter a non-proliferative state termed “senescence” [53], characterized by enhanced glucose uptake and lactate efflux [54]. Here, fibroblasts acquire a senescence-associated secretory phenotype (SASP), depicted by a strong release of IL6, IL1, IL8, GROα and GROβ [55], which contributes to the generation of a tumor-promoting microenvironment [37]. Notably, a tumor cell senescence reinforcing-effect has been assessed on fibroblasts in response to release of TGF-β by recruited macrophages [56].
2.3. Does the TGF-β Dependent CAF Activity Shape the Tumor Metabolism?
Beside the TGF-β-dependent metabolic alteration of fibroblasts, responsible for CAF generation, TGF-β also triggers critical CAF metabolic functions in cancer [32]. Indeed, it has been proposed that, similarly to the Warburg effect in cancer cells, CAFs participate to the metabolic programming of the TME that further nourishes tumor-development [57]. In this scenario, cancer cells and CAFs become metabolically associated [58]. The metabolic crosstalk between stromal and cancer cells, where CAF-generated metabolites sustain the tumor metabolic requirement, takes the name of “reverse Warburg effect”. Here, cancer cells release hydrogen peroxide into the microenvironment, thus fostering oxidative stress in the adjacent CAFs. Concomitantly, CAFs undergo aerobic glycolysis and generate high level of energy-rich fuels (as lactate, pyruvate, ketone bodies and fatty acids), that feed cancer cells [57]. More insight, lactate is secreted by CAF through the monocarboxylate transporter 4 (MCT4), together with an H+ efflux. Therefore, a pH changes responsible for the EMT increase and MMP activity occurs in the TME [59]. In addition, monocarboxylate transporter 1 (MCT1) catalyzes the rapid transport of CAF-generated lactate across the plasma membranes of tumor cells [60]. Here, the concurrent uptake of CAF-released erythrose-4-phosphate leads to a sustained pyruvate production exploited as fuel for pentose phosphate pathway (PPP) [61]. Moreover, multiple ketone bodies accumulated from the CAF glycolytic metabolism, such as acetoacetate and β-hydroxybutyrate, provide a precious tumor energy source for Acetyl-CoA acetyltransferase and 3-hydroxy-3-methylglutaryl-CoA synthase activity during the ketone recycle [58][62]. The metabolic reprogramming in CAFs also induces a high release of glutamine used by tumor cells as nitrogen donor for amino acid synthesis [63]. Finally, CAF-related oxidative stress contributes to ROS accumulation within the tumor, thus supporting HIF and NFκB activation [64].
3. TGF-β Induced Endothelial Metabolic Reprogramming
3.1. Endothelial Cell (EC) General Function
Blood vessels are crucial for the oxygen and nutrient transport in the organism that enables a proper development and maintenance of the perivascular tissue homeostasis [65]. Furthermore, the vascular network plays an indispensable role in draining metabolic waste and, upon request, activate thrombotic and inflammatory responses [66]. During the adulthood, new branches are mostly generated by the tightly cooperated action of leading tip cells, that migrate into avascular region, and the stalk cells, that proliferate to follow the moving tip cells [67]. Once newly formed sprouts meet each other, they fuse to form a new vessel. Here, tip and stalk cells differentiate into the so called phalanx cells, and then the maturation of the vessel occurs with the recruitment of mainly pericytes an fibroblasts [68]. Within the tumor, excessive microvascular network is formed to supply the high request of a growing tumor mass characterized by a chronically active pro-angiogenic signature [69]. However, cancer-associated vessels often show structural and functional abnormalities. The tumor vasculature lacks the organized hierarchy of a normal vascular bed and instead comprises leaky, tortuous, and immature branches [70]. Therefore, the blood flow results chaotic and slow with a consequent inefficient cancer perfusion finally conveying the insurgence of persisting or intermittent hypoxic areas [69][71]. Accordingly, the TME becomes subjected to HIF that drives transcriptional responses promoting adaptation and selection of both cancer and stromal cells, thus inducing pro-survival changes [72]. In this scenario, EC metabolic alterations crucially affect TME composition and function. Here, we will focus our attention on relevant TGF-β mediated processes involving ECs.
3.2. How Does TGF-β Directly Alter EC Metabolic Reprogramming?
ECs demonstrate a high plasticity, with several tunable and dynamic phenotypes switching from Tip, Stalk and Phalanx cells. Beside different names, these multiple functions reflect different proliferative and biosynthetic requirements associated with precise metabolic state and demands [1]. In migrating tip cells, for instance, the rapid energy supply required for fast moving is guaranteed by high glycolytic flux which can produce more ATP in a shorter time with respect to oxidative metabolism [73]. Here, 85% of the total cellular ATP content relays to glycolysis [74]. Indeed, upon pro-angiogenic factor stimulation, GLUT1 and glycolysis regulator 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), an activator of phosphofructokinase 1, are upregulated at lamellipodia and filopodia with the consequent increased in glycolytic flux [74]. Concomitantly, the pro-stalk signaling Dll4 starts to be highly expressed on tip cells. Dll4 activates Notch signaling on the near stalk cells prompting their differentiation and delineating their metabolism. Indeed, the Dll4-dependent cleavage of the Notch intracellular domain (NICD) transcriptionally downregulates the PFKFB3-driven glycolysis and conversely enhances fatty acid (FA) binding proteins (FABP) expression [74]. FABP-dependent FA metabolism is essential for EC proliferation, a key function of stalk cells. Notably, FAs are loaded into stalk cells by a “flip-flop” mechanism or through specific transport proteins, such as CD36 and FA transport proteins [75]. The rate-controlling step of stalk cell FA β-oxidation (FAO) depends on the activity of the carnitine palmitoyltransferases (CPTs) that shuttles the activated fatty-acyl-CoA synthase (FA-CoA) to mitochondria. This process replenishes the tricarboxylic acid (TCA) cycle and leads to the production of aspartate as a precursor for deoxyribonucleotide triphosphate (dNTPs) for cell replication [76]. Despite being deeply characterized for tip and stalk cells, phalanx cell metabolism is still poorly elucidated. However, a central role has been recently given to FOXO1 that belongs to the forkhead box O (FOXO) proteins, a sub-group of the FOX transcription factor family [77]. FOXO1 acts inhibiting Myc, a key transcriptional factor in growth and anabolic metabolism, to decrease glycolysis and impair mitochondrial function, thus ultimately inducing EC quiescence [78]. Acetyl-CoA produced by FAO enters the TCA cycle, with the consequent antioxidative NADPH for the maintenance of barrier integrity. In this line, oxidative pentose phosphate pathway (oxPPP) produces additional NADPH exploitable to regenerate antioxidant reduced glutathione (GSH) [66]. TGF-β and BMP pathways both have effects on angiogenesis through the ALK-1 interaction, which is specifically expressed in vascular EC. They can bind to ALK-1 activating the EC proliferation via pSmad1/5 signaling [79]. Conversely, different endothelial activation states reflect changing in endothelial TGF-β release. In particular, it has been reported that sprouting endothelial tips are characterized by reduced TSP-1 transcription and enhanced expression of pro-tumor factors POSTN and TGF-β1 [80]. By contrast, a stable microvasculature constitutes a dormant niche described by high level of TSP-1, a potent regulator of latent TGF-β activation [81]. Beside the conventional states, EC can be triggered toward an endothelial-to-mesenchymal transition (EndoMT) under TGF-β control during tumor progression [82]. This process involves the activation of a trans-differentiation program whereby ECs escape their endothelial characteristics and gain mesenchymal behavior. EndoMT promotes cancer development and metastasis, but also influence the response to cancer therapy [83]. Indeed, the EndoMT cytoskeletal reorganization through the Rho/ROCK signaling pathway linked to the loss of endothelial adhesion molecules (VE-cadherin, claudins) contributes to the disruption of the endothelial barrier favoring the intra- and extravasation of tumor cells [82]. Furthermore, the aberrant vessel maturation, the decreased expression of vascular endothelial growth factor receptor (VEGFR) and the hypoxia promotion concur to vanish anti-angiogenic strategies and confer resistance to anti-tumoral therapies [82].
During EndoMT, EC undergo morphological, functional and molecular changes, including a decline of their adhesion molecules, increase expression of mesenchymal biomarkers, and conversion of their apico-basal polarity in favor of a front-end back polarity. TGF-β induces EndoMT alterations by involving the Snail family of transcription factors such as Snail, Slug, Twist, and ZEB [84]. Snail family activation can occur through: (i) the direct action of TGF-β involving both Smad-dependent and PI3K/p38 MAPK-dependent signaling pathways [85][86] and (ii) through the hypoxia mediated phosphorylation of the transcription factor Twist-1 [87]. Moreover, activation of the EndoMT program has been recently associated with a metabolic alteration. Indeed, a decreased expression of CPT1 and a decline of acetyl-CoA levels conduits to both the inhibition of FAO, and the associated EndoMT [88]. Again, TGF-β participates to this metabolic-driven EndoMT response by impairing SMAD7 acetylation and signaling [88][89].
3.3. Does the EC Activity Shape the Tumor Metabolism?
As described, multiple events altering EC metabolism in tumors crucially impinge on cancer fate. It has to be considered that cancer cells and ECs reciprocally stimulate each other’s behavior. Indeed, whether angiogenesis occurs to promptly feed the metabolically demanding cancer cells, these latter enhance metabolism of ECs through releasing pro-angiogenic factors. The establishment of this positive feed-forward loop promotes both vessel growth and tumor development [90]. The constant secretion of pro-angiogenic/pro-glycolytic growth factors such as vascular endothelial growth factor (VEGF) by tumor cells in combination with a hypoxic microenvironment generated by both tumor and stromal cells, modify the EC expression of glycolytic enzymes. Indeed, ECs display higher rates of glycolysis than healthy ECs. Notably, pro-inflammatory cytokines further sustain this process. In response to cytokine activation, such as interleukin (IL)-1β stimulation, IκBα becomes phosphorylated before degradation, resulting in release and activation of p65 and p50. Thus, a cytokine-mediated signaling via NF-κB causes the release of pro-angiogenic factors by cancer, stromal and immune cells [91]. All these pathways participate to the upregulation of genes involved in pyruvate metabolism and glucose transport in hypoxic ECs. In addition to the direct effect of cytokines and hypoxia on ECs, tumor cell metabolism directly reprograms the metabolic pathways of ECs. Indeed, cancer-catalyzed lactate that accumulates in the stromal microenvironment might be internalized by ECs sustaining pyruvate oxidation [92]. Moreover, once uptaken, tumor-derived lactate induces a ROS-mediated NF-κB and IL-8 induction in hypoxic ECs [93]. Furthermore, lactate influx seems to participate in modulating receptor tyrosine kinase expression (among them also VEGFR2) [94] and in PHD2/VHL-dependent degradation [95] thus finally amplifying the VEGF signaling. Beside the direct and indirect effects of tumor cell on EC metabolism, tumor-associated macrophages (TAMs) can also influence EC glycolysis. Indeed, hypoxic TAMs strongly upregulate the expression of REDD1, a negative regulator of mTOR. Consequently, REDD1-mediated mTOR inhibition blocks glycolysis in TAMs avoiding the metabolic competition with ECs. The accumulation of glycolytic substrates, uniquely exploitable by highly demanding ECs sustains the excessive angiogenic responses, with consequent formation of abnormal blood vessels [96].
References
- Lau, A.N.; Vander Heiden, M.G. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40.
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129.
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530.
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200.
- Tong, X.; Zhao, F.; Thompson, C.B. The Molecular Determinants of de Novo Nucleotide Biosynthesis in Cancer Cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37.
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of Hypoxia-Inducible Factor 1alpha in Common Human Cancers and Their Metastases. Cancer Res. 1999, 59, 5830–5835.
- Dang, C.V. C-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11.
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell 2018, 33, 368–385.e7.
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230.
- David, C.J.; Massagué, J. Contextual Determinants of TGFβ Action in Development, Immunity and Cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435.
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82.
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β Structure and Activation. Nature 2011, 474, 343–349.
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.F.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 Is a Major Activator of TGF-Β1 In Vivo. Cell 1998, 93, 1159–1170.
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) Is Essential for the Surface Expression of Latent TGF-Beta on Platelets and Activated FOXP3+ Regulatory T Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450.
- Qin, Y.; Garrison, B.S.; Ma, W.; Wang, R.; Jiang, A.; Li, J.; Mistry, M.; Bronson, R.T.; Santoro, D.; Franco, C.; et al. A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System. Cell 2018, 174, 156–171.e16.
- Kobayashi, T.; Kim, H.; Liu, X.; Sugiura, H.; Kohyama, T.; Fang, Q.; Wen, F.-Q.; Abe, S.; Wang, X.; Atkinson, J.J.; et al. Matrix Metalloproteinase-9 Activates TGF-β and Stimulates Fibroblast Contraction of Collagen Gels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L1006–L1015.
- Zhu, H.J.; Sizeland, A.M. A Pivotal Role for the Transmembrane Domain in Transforming Growth Factor-Beta Receptor Activation. J. Biol. Chem. 1999, 274, 11773–11781.
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan Presents Ligand to the TGFβ Signaling Receptor. Cell 1993, 73, 1435–1444.
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061.
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural Determinants of Smad Function in TGF-β Signaling. Trends Biochem. Sci. 2015, 40, 296–308.
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139.
- Edlund, S.; Landström, M.; Heldin, C.-H.; Aspenström, P. Transforming Growth Factor-Beta-Induced Mobilization of Actin Cytoskeleton Requires Signaling by Small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914.
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71.
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 Transcriptional Repressor Complex Promotes TGF-Beta Mediated Epithelial-Mesenchymal Transition. Nat. Cell Biol. 2009, 11, 943–950.
- Lamouille, S.; Derynck, R. Cell Size and Invasion in TGF-β–Induced Epithelial to Mesenchymal Transition Is Regulated by Activation of the MTOR Pathway. J. Cell Biol. 2007, 178, 437–451.
- Harvey, R.F.; Pöyry, T.A.A.; Stoneley, M.; Willis, A.E. Signaling from MTOR to EIF2α Mediates Cell Migration in Response to the Chemotherapeutic Doxorubicin. Sci. Signal. 2019, 12, eaaw6763.
- Chiarugi, P.; Cirri, P. Metabolic Exchanges within Tumor Microenvironment. Cancer Lett. 2016, 380, 272–280.
- Cirri, P.; Chiarugi, P. Cancer Associated Fibroblasts: The Dark Side of the Coin. Am. J. Cancer Res. 2011, 1, 482–497.
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of Fibroblast Heterogeneity in the Tumor Microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646.
- Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers 2019, 11, 619.
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Nathan, S.S.; Huvos, A.G.; Casas-Ganem, J.E.; Yang, R.; Linkov, I.; Sowers, R.; DiResta, G.R.; Gorlick, R.; Healey, J.H. Tumor Interstitial Fluid Pressure May Regulate Angiogenic Factors in Osteosarcoma. J. Orthop. Res. 2008, 26, 1520–1525.
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and Biochemical Remodeling of Stromal Extracellular Matrix in Cancer. Trends Biotechnol. 2015, 33, 230–236.
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 2009, 69, 4167–4174.
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835.
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and Its Microenvironment: A Synergistic Interplay. Semin. Cancer Biol. 2013, 23, 522–532.
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF Beta1 Induces Prolonged Mitochondrial ROS Generation through Decreased Complex IV Activity with Senescent Arrest in Mv1Lu Cells. Oncogene 2005, 24, 1895–1903.
- Byun, H.-O.; Jung, H.-J.; Seo, Y.-H.; Lee, Y.-K.; Hwang, S.-C.; Hwang, E.S.; Yoon, G. GSK3 Inactivation Is Involved in Mitochondrial Complex IV Defect in Transforming Growth Factor (TGF) Β1-Induced Senescence. Exp. Cell Res. 2012, 318, 1808–1819.
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-β Regulates Nox4, MnSOD and Catalase Expression, and IL-6 Release in Airway Smooth Muscle Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 300, L295–L304.
- Liu, R.-M.; Vayalil, P.K.; Ballinger, C.; Dickinson, D.A.; Huang, W.-T.; Wang, S.; Kavanagh, T.J.; Matthews, Q.L.; Postlethwait, E.M. Transforming Growth Factor β Suppresses Glutamate-Cysteine Ligase Gene Expression and Induces Oxidative Stress in A Lung Fibrosis Model. Free Radic. Biol. Med. 2012, 53, 554–563.
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47.
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. BioMed Res. Int. 2018, 2018, 6075403.
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor Cells Induce the Cancer Associated Fibroblast Phenotype via Caveolin-1 Degradation: Implications for Breast Cancer and DCIS Therapy with Autophagy Inhibitors. Cell Cycle 2010, 9, 2423–2433.
- Hou, X.; Zhang, J.; Wang, Y.; Xiong, W.; Mi, J. TGFBR-IDH1-Cav1 Axis Promotes TGF-β Signalling in Cancer-Associated Fibroblast. Oncotarget 2017, 8, 83962–83974.
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and Cancer Metabolism in the Tumor Microenvironment: Markers, Models, and Mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467.
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 Is A Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS ONE 2014, 9, e87871.
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.; Lin, Z.; Witkiewicz, A.; et al. Oxidative Stress in Cancer Associated Fibroblasts Drives Tumor-Stroma Co-Evolution. Cell Cycle 2010, 9, 3276–3296.
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and Mitochondrial SOD2 (MnSOD) Function as Tumor Suppressors in the Stromal Microenvironment: A New Genetically Tractable Model for Human Cancer Associated Fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394.
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of Oxidative Stress and the Microenvironment in Breast Cancer Development and Progression. Adv. Cancer Res. 2013, 119, 107–125.
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-Beta in CAF-Mediated Tumor Growth and Metastasis. Semin. Cancer Biol. 2014, 25, 15–22.
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and Senescence in Cancer-Associated Fibroblasts Metabolically Supports Tumor Growth and Metastasis via Glycolysis and Ketone Production. Cell Cycle 2012, 11, 2285–2302.
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent Stroma Promotes Prostate Cancer Progression: The Role of MiR-210. Mol. Oncol. 2014, 8, 1729–1746.
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Nat. Cell Biol. 2019, 21, 94–101.
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal–Epithelial Metabolic Coupling in Cancer: Integrating Autophagy and Metabolism in the Tumor Microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051.
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The Reverse Warburg Effect Is Likely to Be an Achilles’ Heel of Cancer That Can Be Exploited for Cancer Therapy. Oncotarget 2017, 8, 57813–57825.
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate Transporter 4 (MCT4) Is a High Affinity Transporter Capable of Exporting Lactate in High-Lactate Microenvironments. J. Biol. Chem. 2019, 294, 20135–20147.
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernández-Pérez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-Cell-Autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865.e6.
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701.
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone Bodies and Two-Compartment Tumor Metabolism: Stromal Ketone Production Fuels Mitochondrial Biogenesis in Epithelial Cancer Cells. Cell Cycle 2012, 11, 3956–3963.
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700.
- LaGory, E.L.; Giaccia, A.J. The Ever Expanding Role of HIF in Tumour and Stromal Biology. Nat. Cell Biol. 2016, 18, 356–365.
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307.
- Dumas, S.J.; García-Caballero, M.; Carmeliet, P. Metabolic Signatures of Distinct Endothelial Phenotypes. Trends Endocrinol. Metab. 2020, 31, 580–595.
- Betz, C.; Lenard, A.; Belting, H.-G.; Affolter, M. Cell Behaviors and Dynamics during Angiogenesis. Development 2016, 143, 2249–2260.
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.-M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851.
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Bennewith, K.L.; Durand, R.E. Quantifying Transient Hypoxia in Human Tumor Xenografts by Flow Cytometry. Cancer Res. 2004, 64, 6183–6189.
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The Hypoxic Tumour Microenvironment. Oncogenesis 2018, 7, 1–13.
- Zheng, J. Energy Metabolism of Cancer: Glycolysis versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157.
- Zecchin, A.; Kalucka, J.; Dubois, C.; Carmeliet, P. How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Front. Immunol. 2017, 8, 1750.
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol. Rev. 2010, 90, 367–417.
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis Revisited from a Metabolic Perspective: Role and Therapeutic Implications of Endothelial Cell Metabolism. Open Biol. 2017, 7, 170219.
- Coomans de Brachène, A.; Demoulin, J.-B. FOXO Transcription Factors in Cancer Development and Therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172.
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 Couples Metabolic Activity and Growth State in the Vascular Endothelium. Nature 2016, 529, 216–220.
- Ning, J.; Zhao, Y.; Ye, Y.; Yu, J. Opposing Roles and Potential Antagonistic Mechanism between TGF-β and BMP Pathways: Implications for Cancer Progression. EBioMedicine 2019, 41, 702–710.
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The Perivascular Niche Regulates Breast Tumor Dormancy. Nat. Cell Biol. 2013, 15, 807–817.
- Prunier, C.; Baker, D.; ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78.
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-Mesenchymal Transition in Cancer. Front. Cell Dev. Biol. 2020, 8, doi:10.3389/fcell.2020.00747.
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260.
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324.
- Medici, D.; Potenta, S.; Kalluri, R. Transforming Growth Factor-Β2 Promotes Snail-Mediated Endothelial–Mesenchymal Transition through Convergence of Smad-Dependent and Smad-Independent Signalling. Biochem. J. 2011, 437, 515–520.
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail Is Required for TGFβ-Induced Endothelial-Mesenchymal Transition of Embryonic Stem Cell-Derived Endothelial Cells. J. Cell Sci. 2008, 121, 3317–3324.
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-Induced Pulmonary Hypertension through Transforming Growth Factor-β–Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2017, 58, 194–207.
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.-X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7.
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.-S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294.
- Verdegem, D.; Moens, S.; Stapor, P.; Carmeliet, P. Endothelial Cell Metabolism: Parallels and Divergences with Cancer Cell Metabolism. Cancer Metab. 2014, 2, 19.
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985.
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease: Impact of Hypoxia. EMBO J. 2017, 36, 2187–2203.
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-ΚB/IL-8 Pathway That Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560.
- Ruan, G.-X.; Kazlauskas, A. Lactate Engages Receptor Tyrosine Kinases Axl, Tie2, and Vascular Endothelial Growth Factor Receptor 2 to Activate Phosphoinositide 3-Kinase/Akt and Promote Angiogenesis. J. Biol. Chem. 2013, 288, 21161–21172.
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-M.; Kang, M.; Jang, Y.J.; Yang, S.-J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609.
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715.

