 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lawrence I. Grossman | + 1706 word(s) | 1706 | 2021-02-20 04:16:14 | | | |
| 2 | Camila Xu | Meta information modification | 1706 | 2021-02-25 10:43:26 | | |
Video Upload Options
Complex IV, or cytochrome c oxidase (COX), is the terminal and probably rate-limiting enzyme of the electron transport chain, responsible for accepting electrons from cytochrome c, pumping protons to contribute to the gradient utilized by ATP synthase to produce ATP, and reducing oxygen to water. As such, COX is tightly regulated through numerous mechanisms including protein–protein interactions.
1. Introduction
Mitochondria are the major source of cellular energy that is required to sustain life. They are double-membrane organelles in which the process of cellular respiration and ATP production takes place. This process, oxidative phosphorylation, occurs at the electron transport chain (ETC), a series of four protein complexes embedded in the inner mitochondrial membrane (IM). The complexes create a proton gradient by pumping protons from the matrix to the intermembrane space (IMS), which is coupled with electron transfer down the chain. The electrochemical proton gradient thereby produced is used by ATP synthase (complex V) to generate ATP from ADP and phosphate.
Complex IV, or cytochrome c oxidase (COX), is the terminal enzyme of the ETC and is responsible for reducing oxygen to water. Physiologically, the mammalian complex is a dimer, with each monomer composed of 13 tightly bound subunits embedded in the IM, an assembly supported by several crystal structures resolved from COX in bovine heart [1][2]. However, more recently, monomeric crystal structures of COX were also published [3][4] and monomeric COX was also reported in a supercomplex [5]. It is therefore possible that an equilibrium exists between dimeric and monomeric COX, which could be subject to regulation. In addition, a 14th subunit has been proposed—NDUFA4—which was originally believed to be a subunit of complex I [6][7]. A structural study showed that NDUFA4 appears to be a subunit in the COX monomer, likely adding to the stability of the complex [7]. NDUFA4 as part of the COX monomer is located at the interface of the dimeric complex, where it would prevent or interfere with dimer formation and which could be a reason that the protein was never detected in the dimeric crystal structure. The validity of NDUFA4′s role as a true subunit has been questioned and it was argued that, because NDUFA4 may bind to both complexes I and IV and is not consistently found in COX preparations, it may function as an assembly factor for the respirasome [8].
The three largest subunits are encoded by the mitochondrial genome whereas the other subunits are encoded by the nuclear genome. Among the mitochondrial-encoded subunits, subunits I and II contain the catalytic centers. The latter consist of metal centers that are involved in the electron acceptance from complex III via cytochrome c and the pathway of the electron through the complex itself: electrons received from cytochrome c first reach the CuA center in subunit II, are then transferred to heme a in subunit I, and finally reach the heme a3-CuB site of subunit I, where oxygen is reduced to water.
There are various modes of regulation of COX activity [1], summarized in Table 1. The purpose of this review is to explore the regulation of COX through the interaction with proteins of the twin CX9C family. Members of this protein family have been shown to be important in COX complex assembly and function, as well as for direct regulation of the oxidase [9] (Table 2). Note that the 13 tightly bound COX subunits are traditionally distinguished by Roman numerals introduced by the Kadenbach lab, whereas auxiliary proteins are designated with Arabic numerals (yeast nomenclature can be found in Table 2).
Table 1. COX regulation.
| Types of Regulation of COX |
|---|
| Expression of tissue-, developmental-, and/or species-specific isoforms of subunits |
| Interaction with small molecules |
| Reversible phosphorylation of subunits |
| Protein–protein interactions |
| Supercomplex formation |
Table 2. Human and yeast proteins, nomenclature, and functions of twin CX9C proteins with COX [9].
| Human Protein | Yeast Protein | Function |
|---|---|---|
| CX9C Proteins | ||
| COX17 | Cox17p | COX copper chaperone |
| COX19 | Cox19p | COX assembly |
| CMC1 | Cmc1p | COX assembly |
| CMC2 | Cmc2p | COX assembly |
| COA5 | Pet191p | COX assembly |
| COA6 | Coa6p | COX assembly |
| CHCHD7 | Cox23p | COX assembly |
| CHCHD8 | Coa4p | COX/complex III assembly/function |
| MNRR1/CHCHD2 | Mix17p | Activity regulation |
| CHCHD10 | Mix17p | Activity regulation |
| CMC4 | Cmc4p | Unknown |
| COX VIb1 | Cox12p | Subunit |
| Cytochrome c Oxidase | ||
| COX I | Cox1p | Subunit |
| COX II | Cox2p | |
| COX III | Cox3p | |
| COX IV | Cox4p | |
| COX Va | Cox5Ap | |
| COX Vb | Cox5Bp | |
| COX VI | Cox6p | |
| COX VII | Cox7p | |
| COX VIII | COX8p | |
| COX IX | Cox9p | |
| COX XIII | Cox13p | |
The twin CX9C family of proteins is characterized by its unique motif of two cysteines separated by usually nine amino acid residues. This motif is found in the coiled-coil-helix-coiled-coil-helix (CHCH) domain, where pairs of cysteines form a helix turn helix fold by forming disulfide bonds with one another [10][11][12]. Another family of proteins, called the “small Tim” proteins, contains a similar but shorter twin CX3C motif and plays chaperone roles in the TIM22 pathway for insertion of proteins into the IMS-facing side of the inner membrane (IM) [13]. The CHCH domain is important for the import of the proteins into the intermembrane space (IMS) of the mitochondria. IMS import is facilitated through the Mia40/CHCHD4 redox mechanism [14][15]. The first studies of this family of proteins took place in Saccharomyces cerevisiae, where a detailed study found that 13 of the 14 yeast family members were conserved across species [16]. A follow-up study contained a genome-wide analysis to determine family member functions, with six of the CX9C proteins determined to be involved in COX assembly [9]. Recently, more information has become available through further research into the function of twin CX9C family members.
2. COX Regulation through Assembly
The biogenesis and maturation of COX is critical for its proper function. There are multiple steps in this tightly regulated process: the insertion of metal groups in COX I and COX II, the import and folding of nuclear encoded subunits, and the proper assembly of the subunits into the complex. Over 30 auxiliary proteins are involved in the biogenesis of the core enzyme composed of COX I, COX II, and COX III [17]. The hypothesized assembly pathway favors a modular–linear assembly, where subunits are first assembled into module intermediates and then these modules are assembled into the COX monomer (Figure 1) [18][19]. The first step of monomer assembly is the synthesis of mitochondrially encoded COX I, including the insertion of heme a, which is followed by its association with the COX IV and COX Va module [18][20]. The COX II module, which requires the insertion of the CuA center into the subunit before assembly can continue [21][22][23][24], forms a complex intermediate with COX VIc, COX VIIb, COX VIIc, and COX VIIIa. The COX III module consists of COX III, COX VIa, COX VIb, and COX VIIa. These modules are then assembled in a linear fashion, upon which NDUFA4 interacts to assist in the stabilization of the COX monomer [18]. Detailed COX biogenesis and assembly has been reviewed elsewhere [19]; however, summaries of assembly will be provided where necessary.
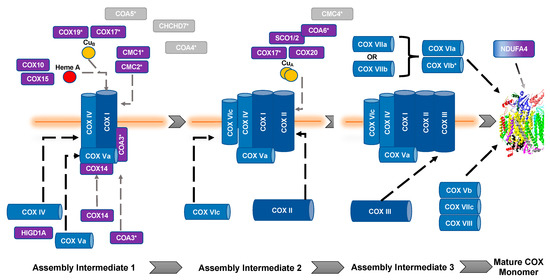
Figure 1. Assembly of cytochrome c oxidase. The assembly of COX is thought to be both modular and linear. Dark blue, mitochondrial encoded COX core subunits; light blue, nuclear encoded COX subunit; purple, assembly factors; gray, unknown function. *, Twin CX9C protein. Gray dashed lines represent assembly factor interactions. Black dashed lines represent subunit assembly.
It is important to note that copper metabolism and homeostasis in the mitochondria are important in aerobic respiration of the cell (for detailed reviews, see [25][26]). All of the assembly proteins discussed in this section have been shown to be involved or associated with copper transport and moiety insertion into appropriate subunits during COX assembly, which is an absolute necessity for the complex to function. Chaperones are very important in this process because of the redox sensitivity of the transition metal that, if left unchecked, can become a source of ROS (for review, see [27]).
Most of the twin CX9C proteins involved in COX assembly were first identified in yeast in a screen to identify proteins that affect OXPHOS; the proteins that were shown to affect OXPHOS were COX17, COX23, COX19, CMC1, and CMC2 [16]. COA4 and COA6 were identified in separate studies [28][29].
3. COX Structural Subunits
COX VIb1/Cox12p
Although not a true twin CX9C protein, this protein is often considered an “unofficial” family member in the context of COX due to structural similarity and its role in complex IV function. The nuclear encoded Cox12p/COX VIb1 has one CX9C motif and one CX10C motif. Cox12p in yeast was first shown to be a novel subunit that is required for proper COX activity but not complex assembly [30]. Cox12p protein is associated specifically with COX II biogenesis, where interaction studies show its physical interaction with COX II, COA6, and SCO1/2 [31]. The COX6B gene was mapped in human heart and three pseudogenes were identified and subsequently characterized [32][33]. COX6B1 is ubiquitously expressed across tissues whereas its isoform, COX6B2, is testes-specific [34]. COX VIb1 shares 45% sequence identity with Cox12p (Figure 10). In 1997, the dimeric complex of COX was crystallized from bovine heart and its structure determination confirmed that COX VIb1 is part of the COX complex, sitting on top of the dimer and seemingly forming a bridge on the IMS side of COX that allows the COX monomers to interact [2]. The crystal structure shows that COX VIb1 interacts with core subunits II and III [2] and, based on computer modeling [35], is part of the cytochrome c binding site of COX.
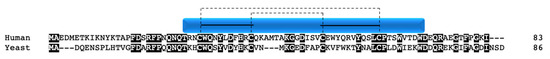
Figure 10. COX VIb1/Cox12p. Alignment of human and yeast COX VIb1/Cox12p protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Some mutations in COX6B1 have been associated with COX deficiency disease. Two individuals with early-onset leukodystrophic encephalopathy, myopathy, and growth retardation carried a homozygous mutation in COX6B1 resulting in an amino acid residue replacement of arginine to histidine at the N-terminus of the protein [36]. Another missense mutation, resulting in an arginine to cysteine change at the same position, was mapped in an individual with encephalomyopathy, hydrocephaly, and cardiomyopathy [37].
References
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76, doi:10.1016/j.mito.2015.07.002.
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 1996, 272, 1136.
- Osuda, Y.; Shinzawa-Itoh, K.; Tani, K.; Maeda, S.; Yoshikawa, S.; Tsukihara, T.; Gerle, C. Two-dimensional crystallization of monomeric bovine cytochrome c oxidase with bound cytochrome c in reconstituted lipid membranes. Microsc. (Oxf) 2016, 65, 263–267, doi:10.1093/jmicro/dfv381.
- Shinzawa-Itoh, K.; Sugimura, T.; Misaki, T.; Tadehara, Y.; Yamamoto, S.; Hanada, M.; Yano, N.; Nakagawa, T.; Uene, S.; Yamada, T., et. al. Monomeric structure of an active form of bovine cytochrome c oxidase. Proc Natl Acad Sci U S A 2019, 116, 19945–19951, doi:10.1073/pnas.1907183116.
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of mammalian respiratory supercomplex I2III2IV2. Cell 2016, 167, 1598–1609 e1510, doi:10.1016/j.cell.2016.11.012.
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab 2012, 16, 378–386, doi:10.1016/j.cmet.2012.07.015.
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res 2018, 28, 1026–1034, doi:10.1038/s41422–018–0071–1.
- Kadenbach, B. Regulation of mammalian 13-subunit cytochrome c oxidase and binding of other proteins: role of NDUFA4. Trends Endocrinol Metab 2017, 28, 761–770, doi:10.1016/j.tem.2017.09.003.
- Cavallaro, G. Genome-wide analysis of eukaryotic twin CX9C proteins. Mol Biosyst 2010, 6, 2459–2470, doi:10.1039/c0mb00058b.
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J Biol Chem 1996, 271, 14504–14509, doi:10.1074/jbc.271.24.14504.
- Abajian, C.; Yatsunyk La Fau - Ramirez, B.E.; Ramirez Be Fau - Rosenzweig, A.C.; Rosenzweig, A.C. Yeast Cox17 solution structure and Copper(I) binding. J Biol Chem 2004, 279, 53584–53592.
- Arnesano, F.; Balatri, E.; Banci, L.; Bertini, I.; Winge, D.R. Folding studies of Cox17 reveal an important interplay of cysteine oxidation and copper binding. Structure 2005, 13, 713–722, doi:http://dx.doi.org/10.1016/j.str.2005.02.015.
- Koehler, C.M. The small Tim proteins and the twin Cx3C motif. Trends Biochem Sci 2004, 29, 1–4, doi:10.1016/j.tibs.2003.11.003.
- Terziyska, N.; Lutz, T.; Kozany, C.; Mokranjac, D.; Mesecke, N.; Neupert, W.; Herrmann, J.M.; Hell, K. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. Febs Lett 2005, 579, 179–184, doi:10.1016/j.febslet.2004.11.072.
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Sanjuan Szklarz, L.K.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. Embo J 2004, 23, 3735–3746, doi:10.1038/sj.emboj.7600389.
- Longen, S.; Bien, M.; Bihlmaier, K.; Kloeppel, C.; Kauff, F.; Hammermeister, M.; Westermann, B.; Herrmann, J.M.; Riemer, J. Systematic analysis of the twin CX9C protein family. J. Mol. Biol. 2009, 393, 356–368, doi:http://dx.doi.org/10.1016/j.jmb.2009.08.041.
- Barrientos, A.; Barros, M.H.; Valnot, I.; Rotig, A.; Rustin, P.; Tzagoloff, A. Cytochrome oxidase in health and disease. Gene 2002, 286, 53–63, doi:10.1016/s0378–1119(01)00803–4.
- Vidoni, S.; Harbour, M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E., et. al. MR-1S interacts with PET100 and PET117 in module-based assembly of human cytochrome c oxidase. Cell Rep 2017, 18, 1727–1738, doi:10.1016/j.celrep.2017.01.044.
- Timon-Gomez, A.; Nyvltova, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin Cell Dev Biol 2018, 76, 163–178, doi:10.1016/j.semcdb.2017.08.055.
- Stiburek, L.; Vesela, K.; Hansikova, H.; Pecina, P.; Tesarova, M.; Cerna, L.; Houstek, J.; Zeman, J. Tissue-specific cytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1. Biochem J 2005, 392, 625–632, doi:10.1042/BJ20050807.
- Williams, S.L.; Valnot, I.; Rustin, P.; Taanman, J.W. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J Biol Chem 2004, 279, 7462–7469, doi:10.1074/jbc.M309232200.
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.G.; Gerbitz, K.D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum Mol Genet 2000, 9, 795–801, doi:10.1093/hmg/9.5.795.
- Jaksch, M.; Paret, C.; Stucka, R.; Horn, N.; Muller-Hocker, J.; Horvath, R.; Trepesch, N.; Stecker, G.; Freisinger, P.; Thirion, C., et. al. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum Mol Genet 2001, 10, 3025–3035, doi:10.1093/hmg/10.26.3025.
- Leary, S.C.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum Mol Genet 2004, 13, 1839–1848, doi:10.1093/hmg/ddh197.
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. Iubmb Life 2008, 60, 421–429, doi:10.1002/iub.50.
- Leary, S.C.; Winge, D.R.; Cobine, P.A. "Pulling the plug" on cellular copper: the role of mitochondria in copper export. Biochim Biophys Acta 2009, 1793, 146–153, doi:10.1016/j.bbamcr.2008.05.002.
- Valentine, J.S.; Wertz, D.L.; Lyons, T.J.; Liou, L.L.; Goto, J.J.; Gralla, E.B. The dark side of dioxygen biochemistry. Curr Opin Chem Biol 1998, 2, 253–262, doi:10.1016/s1367–5931(98)80067–7.
- Bestwick, M.; Jeong, M.Y.; Khalimonchuk, O.; Kim, H.; Winge, D.R. Analysis of Leigh syndrome mutations in the yeast SURF1 homolog reveals a new member of the cytochrome oxidase assembly factor family. Mol Cell Biol 2010, 30, 4480–4491, doi:10.1128/MCB.00228–10.
- Vogtle, F.N.; Burkhart, J.M.; Rao, S.; Gerbeth, C.; Hinrichs, J.; Martinou, J.C.; Chacinska, A.; Sickmann, A.; Zahedi, R.P.; Meisinger, C. Intermembrane space proteome of yeast mitochondria. Mol Cell Proteom. 2012, 11, 1840–1852, doi:10.1074/mcp.M112.021105.
- LaMarche, A.E.; Abate, M.I.; Chan, S.H.; Trumpower, B.L. Isolation and characterization of COX12, the nuclear gene for a previously unrecognized subunit of Saccharomyces cerevisiae cytochrome c oxidase. J Biol Chem 1992, 267, 22473–22480.
- Ghosh, A.; Pratt, A.T.; Soma, S.; Theriault, S.G.; Griffin, A.T.; Trivedi, P.P.; Gohil, V.M. Mitochondrial disease genes COA6, COX6B and SCO2 have overlapping roles in COX2 biogenesis. Hum Mol Genet 2016, 25, 660–671, doi:10.1093/hmg/ddv503.
- Carrero-Valenzuela, R.D.; Quan, F.; Lightowlers, R.; Kennaway, N.G.; Litt, M.; Forte, M. Human cytochrome c oxidase subunit VIb: characterization and mapping of a multigene family. Gene 1991, 102, 229–236, doi:10.1016/0378–1119(91)90082-m.
- Taanman, J.W.; Schrage, C.; Reuvekamp, P.; Bijl, J.; Hartog, M.; de Vries, H.; Agsteribbe, E. Identification of three human pseudogenes for subunit VIb of cytochrome c oxidase: a molecular record of gene evolution. Gene 1991, 102, 237–244, doi:10.1016/0378–1119(91)90083-n.
- Hüttemann, M.; Jaradat, S.; Grossman, L.I. Cytochrome c oxidase of mammals contains a testes-specific isoform of subunit VIb--the counterpart to testes-specific cytochrome c? Mol Reprod Dev 2003, 66, 8–16, doi:10.1002/mrd.10327.
- Roberts, V.A.; Pique, M.E. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J Biol Chem 1999, 274, 38051–38060, doi:10.1074/jbc.274.53.38051.
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D'Adamo, P.; Gasparini, P.; Zeviani, M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J Hum Genet 2008, 82, 1281–1289, doi:10.1016/j.ajhg.2008.05.002.
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur J Hum Genet 2015, 23, 159–164, doi:10.1038/ejhg.2014.85.


