 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Irena Baranowska-Bosiacka | + 1900 word(s) | 1900 | 2021-02-07 04:05:19 |
Video Upload Options
Hypoxia is an integral component of the tumor microenvironment. Either as chronic or cycling hypoxia, it exerts a similar effect on cancer processes by activating hypoxia-inducible factor-1 (HIF-1) and nuclear factor (NF-κB), with cycling hypoxia showing a stronger proinflammatory influence.
1. Introduction
Growing standards of living and advances in medicine in many countries have resulted in increased life expectancies. This has led to the spread of other diseases, particularly “civilization diseases” which are associated with lifestyle and environmental pollution, and diseases related to old age. One of these is cancer, a major problem in developed countries [1] and the cause of 55.9 million deaths worldwide in 2017 [1][2]—a 25% increase when compared to 2007 [2]. This trend has led to the intensification of efforts aimed at developing more effective therapeutic approaches.
The last 15 years have seen an increasing interest in the tumor microenvironment—a set of factors affecting a cancer cell in a tumor [3]. One of these are noncancer cells recruited to a tumor niche, with the recruitment process depending on factors secreted from cells already located in the tumor niche. These include factors changing the recruited cell into a tumor-associated cell—i.e., transforming the cell into an element which supports cancer processes [4][5]. Both cancer and noncancer cells secrete a number of factors which shape the tumor microenvironment, including chemoattractant cytokines, also known as chemokines [6][7]. Changes in the expression of chemokines affect the recruitment of cells to the tumor niche, angiogenesis and migration of cancer cells. For this reason, understanding the role of chemokines in cancer processes seems crucial for understanding the functioning of a tumor [7].
2. Hypoxia in a Tumor
Tumors are associated with two kinds of hypoxia—chronic hypoxia (also known as continuous or noninterrupted hypoxia) and cycling hypoxia (intermittent or transient hypoxia). Chronic hypoxia is a persistent reduction in oxygen levels that results from an excessive distance between cells and vascular vessels whose growth cannot keep up with the rapid growth of the tumor [8]. Cycling hypoxia has cyclic episodes of oxygen deficiency interspersed with periods of reoxygenation [9]—a result of structural abnormalities in the vascular vessels, particularly the lack of a conventional hierarchy (Figure 1) [10][11]. In such vessels, the blood flow often changes route, resulting in hypoxia in some areas of the tumor. Then, when the blood flow changes its route again, some areas become reoxygenated. One such cycle of hypoxia lasts from several minutes [12] to 4 h [13], depending on the selected research model.
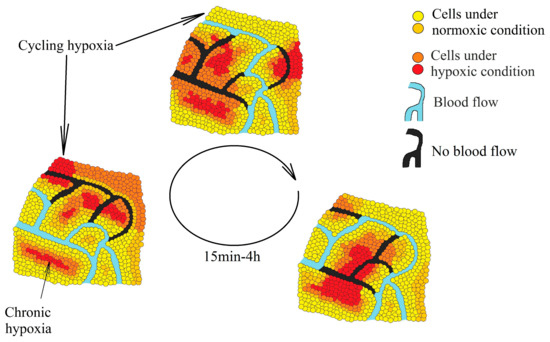
Figure 1. Two types of hypoxia in a tumor: chronic hypoxia associated with an excessive distance from the blood vessels and cycling hypoxia associated with changes in the blood flow in the vessels inside the tumor which lead to periodic oxygen deficiencies in various parts of the tumor.
Both types of hypoxia change the expression of similar genes [14] as they activate the same transcription factors—hypoxia-inducible factors (HIFs) and nuclear factor κB (NF-κB). The mechanisms of activation, however, are different. In chronic hypoxia, the main role is played by a reduction in oxygen levels which triggers a drop in the activity of oxygen-dependent enzymes [15]. In cycling hypoxia, transcription factors are activated mainly by reactive oxygen species (ROS) [16][17].
Hypoxia-inducible factors (HIF-1, HIF-2 and HIF-3) exhibit transcriptive activity that is regulated by the proteolytic degradation of subunit α (HIF-1α and HIF-2α) [18][19][20]. This degradation occurs due to the hydroxylation of HIF-1α and HIF-2α in proline residues by prolyl hydroxylase (PHD), which leads to the ubiquitination by von Hippel-Lindau protein (pVHL) and then to proteolytic degradation of these HIF-α subunits [21]. PHD are oxygen-dependent enzymes (Figure 2) [15]. For this reason, in chronic hypoxia, there is a decrease in PHD activity which leads to a decrease in HIF-α hydroxylation of proline residues and a decrease in proteolytic degradation of these HIF subunits. HIF-1α reaches the maximum level in a cell after 6 h of chronic hypoxia, whereas HIF-2α reaches this after 48 h [22]. The expressions of PHD2 and PHD3 are increased by HIF-1 and HIF-2 [23][24][25][26], which makes HIF-α degrade more intensely during reoxygenation. In turn, cycling hypoxia increases the level of ROS, which oxidizes the iron atoms in PHD and thus reduces the activity of these enzymes [16][27]. This leads to a drop in the hydroxylation of proline residues on HIF-α, which means that HIF-α is no longer degraded but accumulated instead. The increase in HIF-1α levels during cycling hypoxia also occurs via other pathways—for example, the ROS-dependent activation protein kinase A (PKA), which phosphorylates this HIF subunit thereby increasing its stability [28][29]. A ROS-dependent increase in calcium ion concentration in cytoplasm, the activation of protein kinase C (PKC) and the mammalian target of rapamycin (mTOR) are also significant [30]. In cycling hypoxia, the accumulation of HIF-1α increases with the successive cycles of oxygen deficiency [31]. However, this protein is completely degraded during episodes of reoxygenation [31].
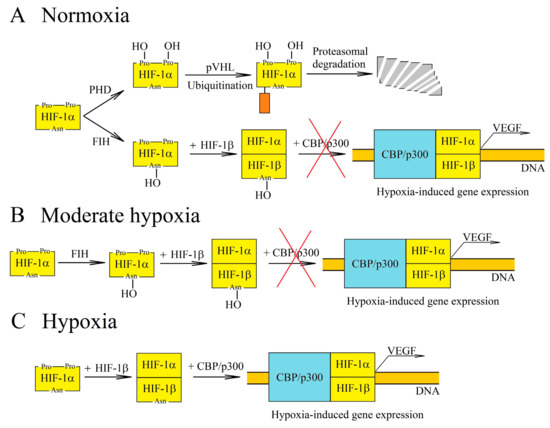
Figure 2. Regulation of hypoxia-inducible factor-1 (HIF-1) transcriptional activity at various oxygen concentrations. (A) In normoxia, HIF-1α is hydroxylated by the oxygen-dependent enzymes prolyl hydroxylase (PHD) and factor inhibiting HIF (FIH). The PHD-catalyzed reaction leads to proteasomal degradation of HIF-1α. In turn, hydroxylation by FIH prevents interaction of HIF-1 with the CBP/p300 coactivator. (B) PHD and FIH require different concentrations of oxygen for their activity and so, in moderate hypoxia, PHD activity decreases while FIH retains its functions. Accumulation of HIF-1α occurs, but due to hydroxylation by FIH, there is no interaction between HIF-1 and the CBP/p300 coactivator, and thus genes dependent on this coactivator are not expressed. (C) In hypoxia, the activities of oxygen-dependent enzymes are reduced. For this reason, HIF-1α is not hydroxylated by PHD or by FIH. This subunit begins to form a transcription factor with HIF-1β, which is responsible for inducing the transcription of hypoxia-dependent genes.
The regulation of HIF transcriptional activity also involves factor inhibiting HIF (FIH), an enzyme which causes the hydroxylation of asparagine residue [32] and thus disrupts the interaction of HIF-α with the CBP/p300 coactivator [33][34]. In this way, FIH blocks the expression of HIF/CBP/p300-dependent genes. As FIH is an oxygen-dependent enzyme [35], its activity is reduced by chronic hypoxia and ROS which oxidize the iron atom—a part of the enzyme that is critical for its function [32]. FIH requires a much lower level of oxygen for its activity than PHD (Km for FIH is 90 μM, whereas for PHD it is 230–250 μM) [15][35][36]. For this reason, in moderate hypoxia, the HIF pathway is inhibited by FIH but not by PHD.
Both types of hypoxia also activate NF-κB. In chronic hypoxia, this activation is dependent, among other things, on a reduction in the amount of oxygen and the ensuing reduction in the activity of PHD, an enzyme that inhibits the activation of NF-κB and HIF [37][38]. The activation of NF-κB in chronic hypoxia also depends on the activation of calcium/calmodulin-dependent kinase 2 (CaMK2) [39]. In contrast, in cycling hypoxia, NF-κB activation is ROS-dependent [17][40][41].
In chronic hypoxia, the activation of NF-κB results in an increased expression of HIF-1α and therefore has a significant effect on the HIF activation pathway [42][43]. This is related to the occurrence of the NF-κB binding site in the HIF1A gene promoter. For this reason, during chronic hypoxia, the expression of genes is directly induced by NF-κB. There is also an increase in the expression of genes directly dependent on HIF-1α but these are also indirectly dependent on NF-κB during chronic hypoxia. Therefore, in order to acquire a detailed insight into the mechanism which induces the expression of a given gene by chronic hypoxia, it is necessary to demonstrate the occurrence and investigate the functionality of the hypoxia responsive element (HRE) binding HIF or the NF-κB binding site. Importantly, chronic hypoxia and inflammation exclude each other by various mechanisms [44][45], and so chronic hypoxia reduces the inflammatory response. On the other hand, some proinflammatory genes are induced by both chronic hypoxia and inflammation [46]. Cycling hypoxia is more proinflammatory than chronic hypoxia [41][47][48]. This is related to the activation of NF-κB by ROS [17][40][41]. For this reason, NF-κB plays a more important role in gene expression during cycling hypoxia than in chronic hypoxia.
Hypoxia significantly changes the functioning of the tumor. Its proapoptotic effect on cells results in a selection of cells in terms of apoptosis resistance, a process which is important at the beginning of tumor development and results in the presence of cancer cells with a p53 dysfunction in the tumor [49]. Hypoxia also participates in the progression of cancer at further stages of the process. In particular, hypoxia is important in the functioning of cancer stem cells (CSCs) [50][51][52][53][54][55], which increase the resistance of the tumor to anticancer therapy. Hypoxia also causes cancer cell migration, invasion and metastasis, partly due to hypoxia causing the epithelial-to-mesenchymal transition (EMT) [55][56][57][58][59]. For this reason, areas of chronic hypoxia are often associated with neoplastic cell metastasis.
Tissues respond to oxygen deficiency by developing new blood vessels. In this way, hypoxia increases the expression of proangiogenic factors such as vascular endothelial growth factor (VEGF)-A [60][61], platelet-derived growth factor subunit A (PDGF-A), transforming growth factor-β (TGF-β) and angiopoietin-like 4 (ANGPTL4) [62]. Hypoxia also participates in tumor immune evasion by polarizing macrophages to the M2 phenotype which silences the immune response [63]. It also protects cancer cells by impairing the function of NK cells [64][65] and increasing the production of immunosuppressive proteins such as indoleamine 2,3-dioxygenase (IDO), human leukocyte antigen-G (HLA-G), programmed death-ligand 1 (PD-L1) and metabolites such as adenosine [66][67]. The hypoxia-induced acidosis of the cancer microenvironment, which is caused by an increased production and secretion of lactate is also important [66][68]. Lactate causes tumor immune evasion and neoplastic cell migration.
Hypoxia also affects the CXC chemokine system, which leads to changes in the level of these chemoattractant cytokines in the cancer microenvironment. CXC chemokines participate in the growth of the tumor due to a number of procancer properties.
HIF-1α accumulation and increased HIF-1 transcriptional activity occurs in cancer cells even in normoxia. This is related to, among other things, mutations in the VHL gene which encodes pVHL, resulting in the loss of biological function of pVHL, thereby reducing the degradation of HIF-1α [69][70]. Tumors also exhibit deletions of parts of the chromosome where the HIF1AN gene locus are located [71]. This gene encodes FIH-1, the enzyme responsible for inhibiting the transcriptional activity of HIF-1. Another way of activating the HIF-1 pathway under normoxia is HIF-1α phosphorylation [20] which leads to the increased stability of this protein and consequently, to HIF-1α accumulation in cells and increased expression of HIF-1dependent genes. Enzymes performing such phosphorylation under normoxia include PKA activated by cAMP [72], phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) [73][74], extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) [75][76][77]. HIF-1α is also phosphorylated by glycogen synthase kinase 3 which reduces the stability of this protein [77]. Apart from phosphorylation, other types of post-translational modifications also affect the activation of HIF-1 under normoxia. One of them is the deacetylation of HIF-1α by sirtuin 1 (SIRT1), which leads to a decrease in HIF-1 transcriptional activity [78]. In addition to the post-translational modification, the activation of NF-κB in normoxia also increases the expression of HIF-1α, which is important for an increase in the activation of HIF-1 during inflammatory reactions [75]. The elevated activation of HIF-1 via these pathways in normoxia increases the expression of genes dependent on this transcriptional factor. Although there are no studies in this area that confirm it, it is possible that this mechanism is responsible for the increase in the expression of CXC chemokines and CXC chemokine receptors in normoxia.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788.
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvi-ronment in tumorigenesis. J. Cancer 2017, 8, 761–773.
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438.
- Lian, G.; Chen, S.; Ouyang, M.; Li, F.; Chen, L.; Yang, J. Colon Cancer Cell Secretes EGF to Promote M2 Polarization of TAM through EGFR/PI3K/AKT/mTOR Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819849068.
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971.
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287.
- Span, P.N.; Bussink, J. Biology of hypoxia. Semin. Nucl. Med. 2015, 45, 101–109.
- Cárdenas-Navia, L.I.; Mace, D.; Richardson, R.A.; Wilson, D.F.; Shan, S.; Dewhirst, M.W. The pervasive presence of fluctuat-ing oxygenation in tumors. Cancer Res. 2008, 68, 5812–5819.
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003, 163, 1801–1815.
- Lanzen, J.; Braun, R.D.; Klitzman, B.; Brizel, D.; Secomb, T.W.; Dewhirst, M.W. Direct demonstration of instabilities in oxygen concentrations within the extravascular compartment of an experimental tumor. Cancer Res. 2006, 66, 2219–2223.
- Panek, R.; Welsh, L.; Baker, L.; Schmidt, M.A.; Wong, K.H.; Riddell, A.M.; Koh, D.M.; Dunlop, A.; Mcquaid, D.; d’Arcy, J.A.; et al. Noninvasive Imaging of Cycling Hypoxia in Head and Neck Cancer Using Intrinsic Susceptibility MRI. Clin. Cancer Res. 2017, 23, 4233–4241.
- Ellingsen, C.; Ovrebø, K.M.; Galappathi, K.; Mathiesen, B.; Rofstad, E.K. pO₂ fluctuation pattern and cycling hypoxia in hu-man cervical carcinoma and melanoma xenografts. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1317–1323.
- Olbryt, M.; Habryka, A.; Student, S.; Jarząb, M.; Tyszkiewicz, T.; Lisowska, K.M. Global gene expression profiling in three tumor cell lines subjected to experimental cycling and chronic hypoxia. PLoS ONE 2014, 9, e105104.
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780.
- Hsieh, C.H.; Lee, C.H.; Liang, J.A.; Yu, C.Y.; Shyu, W.C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636.
- Chen, W.L.; Wang, C.C.; Lin, Y.J.; Wu, C.P.; Hsieh, C.H. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J. Transl. Med. 2015, 13, 389.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978.
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480.
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hy-poxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206.
- Guan, Z.; Ding, C.; Du, Y.; Zhang, K.; Zhu, J.N.; Zhang, T.; He, D.; Xu, S.; Wang, X.; Fan, J. HAF drives the switch of HIF-1α to HIF-2α by activating the NF-κB pathway, leading to malignant behavior of T24 bladder cancer cells. Int. J. Oncol. 2014, 44, 393–402.
- D’Angelo, G.; Duplan, E.; Boyer, N.; Vigne, P.; Frelin, C. Hypoxia up-regulates prolyl hydroxylase activity: A feedback mech-anism that limits HIF-1 responses during reoxygenation. J. Biol. Chem. 2003, 278, 38183–38187.
- Stiehl, D.P.; Wirthner, R.; Köditz, J.; Spielmann, P.; Camenisch, G.; Wenger, R.H. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 2006, 281, 23482–23491.
- Ginouvès, A.; Ilc, K.; Macías, N.; Pouysségur, J.; Berra, E. PHDs overactivation during chronic hypoxia “desensitizes” HI-Falpha and protects cells from necrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4745–4750.
- Fujita, N.; Markova, D.; Anderson, D.G.; Chiba, K.; Toyama, Y.; Shapiro, I.M.; Risbud, M.V. Expression of prolyl hydroxylas-es (PHDs) is selectively controlled by HIF-1 and HIF-2 proteins in nucleus pulposus cells of the intervertebral disc: Distinct roles of PHD2 and PHD3 proteins in controlling HIF-1α activity in hypoxia. J. Biol. Chem. 2012, 287, 16975–16986.
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouysségur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 118, 781–794.
- Toffoli, S.; Feron, O.; Raes, M.; Michiels, C. Intermittent hypoxia changes HIF-1alpha phosphorylation pattern in endothelial cells: Unravelling of a new PKA-dependent regulation of HIF-1alpha. Biochim Biophys. Acta 2007, 1773, 1558–1571.
- Zhang, Y.L.; Tavakoli, H.; Chachisvilis, M. Apparent PKA activity responds to intermittent hypoxia in bone cells: A redox pathway? Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H225–H235.
- Yuan, G.; Nanduri, J.; Khan, S.; Semenza, G.L.; Prabhakar, N.R. Induction of HIF-1alpha expression by intermittent hypoxia: Involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J. Cell Physiol. 2008, 217, 674–685.
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hänze, J. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 2010, 48, 1626–1635.
- Masson, N.; Singleton, R.S.; Sekirnik, R.; Trudgian, D.C.; Ambrose, L.J.; Miranda, M.X.; Tian, Y.M.; Kessler, B.M.; Schofield, C.J.; Ratcliffe, P.J. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012, 13, 251–257.
- Freedman, S.J.; Sun, Z.Y.; Poy, F.; Kung, A.L.; Livingston, D.M.; Wagner, G.; Eck, M.J. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 5367–5372.
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation do-main a hypoxic switch. Science 2002, 295, 858–861.
- Koivunen, P.; Hirsilä, M.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904.
- Tuckerman, J.R.; Zhao, Y.; Hewitson, K.S.; Tian, Y.M.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. Determination and comparison of specific activity of the HIF-prolyl hydroxylases. FEBS Lett. 2004, 576, 145–150.
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159.
- Wang, L.; Niu, Z.; Wang, X.; Li, Z.; Liu, Y.; Luo, F.; Yan, X. PHD2 exerts anti-cancer and anti-inflammatory effects in colon cancer xenografts mice via attenuating NF-κB activity. Life Sci. 2020, 242, 117167.
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-kappaB. Mol. Cell Biol. 2010, 30, 4901–4921.
- Quintero, M.; Gonzalez-Martin, M.D.C.; Vega-Agapito, V.; Gonzalez, C.; Obeso, A.; Farré, R.; Agapito, T.; Yubero, S. The effects of intermittent hypoxia on redox status, NF-κB activation, and plasma lipid levels are dependent on the lowest oxygen saturation. Free Radic. Biol. Med. 2013, 65, 1143–1154.
- Gutsche, K.; Randi, E.B.; Blank, V.; Fink, D.; Wenger, R.H.; Leo, C.; Scholz, C.C. Intermittent hypoxia confers pro-metastatic gene expression selectively through NF-κB in inflammatory breast cancer cells. Free Radic. Biol. Med. 2016, 101, 129–142.
- Belaiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary ar-tery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697.
- Jiang, Y.; Zhu, Y.; Wang, X.; Gong, J.; Hu, C.; Guo, B.; Zhu, B.; Li, Y. Temporal regulation of HIF-1 and NF-κB in hypoxic hepatocarcinoma cells. Oncotarget 2015, 6, 9409–9419.
- Shin, D.H.; Li, S.H.; Yang, S.W.; Lee, B.L.; Lee, M.K.; Park, J.W. Inhibitor of nuclear factor-kappaB alpha derepresses hypox-ia-inducible factor-1 during moderate hypoxia by sequestering factor inhibiting hypoxia-inducible factor from hypoxia-inducible factor 1alpha. FEBS J 2009, 276, 3470–3480.
- Mendonça, D.B.; Mendonça, G.; Aragão, F.J.; Cooper, L.F. NF-κB suppresses HIF-1α response by competing for P300 binding. Biochem. Biophys. Res. Commun. 2011, 404, 997–1003.
- Ravenna, L.; Principessa, L.; Verdina, A.; Salvatori, L.; Russo, M.A.; Petrangeli, E. Distinct phenotypes of human prostate cancer cells associate with different adaptation to hypoxia and pro-inflammatory gene expression. PLoS ONE 2014, 9, e96250.
- Tellier, C.; Desmet, D.; Petit, L.; Finet, L.; Graux, C.; Raes, M.; Feron, O.; Michiels, C. Cycling hypoxia induces a specific am-plified inflammatory phenotype in endothelial cells and enhances tumor-promoting inflammation in vivo. Neoplasia 2015, 17, 66–78.
- Song, D.; Fang, G.; Mao, S.Z.; Ye, X.; Liu, G.; Miller, E.J.; Greenberg, H.; Liu, S.F. Selective inhibition of endothelial NF-κB signaling attenuates chronic intermittent hypoxia-induced atherosclerosis in mice. Atherosclerosis 2018, 270, 68–75.
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91.
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959.
- Seo, E.J.; Kim, D.K.; Jang, I.H.; Choi, E.J.; Shin, S.H.; Lee, S.I.; Kwon, S.M.; Kim, K.H.; Suh, D.S.; Kim, J.H. Hypoxia-NOTCH1-SOX2 signaling is important for maintaining cancer stem cells in ovarian cancer. Oncotarget 2016, 7, 55624–55638.
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056.
- Qin, J.; Liu, Y.; Lu, Y.; Liu, M.; Li, M.; Li, J.; Wu, L. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like proper-ties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 2017, 7, 10592.
- Mahkamova, K.; Latar, N.; Aspinall, S.; Meeson, A. Hypoxia Increases Thyroid Cancer Stem Cell-Enriched Side Population. World J. Surg. 2018, 42, 350–357.
- Bhuria, V.; Xing, J.; Scholta, T.; Bui, K.C.; Nguyen, M.L.T.; Malek, N.P.; Bozko, P.; Plentz, R.R. Hypoxia induced Sonic Hedgehog signaling regulates cancer stemness, epithelial-to-mesenchymal transition and invasion in cholangiocarcinoma. Exp. Cell Res. 2019, 385, 111671.
- Matsuoka, J.; Yashiro, M.; Doi, Y.; Fuyuhiro, Y.; Kato, Y.; Shinto, O.; Noda, S.; Kashiwagi, S.; Aomatsu, N.; Hirakawa, T.; et al. Hypoxia stimulates the EMT of gastric cancer cells through autocrine TGFβ signaling. PLoS ONE 2013, 8, e62310.
- Zuo, J.; Wen, J.; Lei, M.; Wen, M.; Li, S.; Lv, X.; Luo, Z.; Wen, G. Hypoxia promotes the invasion and metastasis of laryngeal cancer cells via EMT. Med. Oncol. 2016, 33, 15.
- Wang, X.H.; He, X.; Jin, H.Y.; Liang, J.X.; Li, N. Effect of hypoxia on the Twist1 in EMT of cervical cancer cells. Eur. Rev Med. Pharmacol. Sci. 2018, 22, 6633–6639.
- Tang, C.; Liu, T.; Wang, K.; Wang, X.; Xu, S.; He, D.; Zeng, J. Transcriptional regulation of FoxM1 by HIF-1α mediates hypox-ia-induced EMT in prostate cancer. Oncol. Rep. 2019, 42, 1307–1318.
- Terashima, J.; Sampei, S.; Iidzuka, M.; Ohsakama, A.; Tachikawa, C.; Satoh, J.; Kudo, K.; Habano, W.; Ozawa, S. VEGF ex-pression is regulated by HIF-1α and ARNT in 3D KYSE-70, esophageal cancer cell spheroids. Cell Biol. Int. 2016, 40, 1187–1194.
- Cesário, J.M.; Brito, R.B.; Malta, C.S.; Silva, C.S.; Matos, Y.S.; Kunz, T.C.; Urbano, J.J.; Oliveira, L.V.; Dalboni, M.A.; Dellê, H. A simple method to induce hypoxia-induced vascular endothelial growth factor-A (VEGF-A) expression in T24 human blad-der cancer cells. In Vitro Cell Dev. Biol. Anim 2017, 53, 272–276.
- Zong, S.; Li, W.; Li, H.; Han, S.; Liu, S.; Shi, Q.; Hou, F. Identification of hypoxia-regulated angiogenic genes in colorectal cancer. Biochem. Biophys. Res. Commun. 2017, 493, 461–467.
- Ke, X.; Chen, C.; Song, Y.; Cai, Q.; Li, J.; Tang, Y.; Han, X.; Qu, W.; Chen, A.; Wang, H.; et al. Hypoxia modifies the polariza-tion of macrophages and their inflammatory microenvironment, and inhibits malignant behavior in cancer cells. Oncol. Lett. 2019, 18, 5871–5878.
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affect-ing ADCC. Eur. J. Immunol. 2013, 43, 2756–2764.
- Chen, C.H.; Li, S.X.; Xiang, L.X.; Mu, H.Q.; Wang, S.B.; Yu, K.Y. HIF-1α induces immune escape of prostate cancer by regu-lating NCR1/NKp46 signaling through miR-224. Biochem. Biophys. Res. Commun. 2018, 503, 228–234.
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front Immunol. 2018, 9, 1591.
- Song, X.; Zhang, Y.; Zhang, L.; Song, W.; Shi, L. Hypoxia enhances indoleamine 2,3-dioxygenase production in dendritic cells. Oncotarget 2018, 9, 11572–11580.
- Pérez-Tomás, R.; Pérez-Guillén, I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244.
- Miller, F.; Kentsis, A.; Osman, R.; Pan, Z.Q. Inactivation of VHL by tumorigenic mutations that disrupt dynamic coupling of the pVHL.hypoxia-inducible transcription factor-1alpha complex. J. Biol. Chem. 2005, 280, 7985–7996.
- Gossage, L.; Eisen, T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat. Rev Clin. Oncol. 2010, 7, 277–288.
- Wang, E.; Zhang, C.; Polavaram, N.; Liu, F.; Wu, G.; Schroeder, M.A.; Lau, J.S.; Mukhopadhyay, D.; Jiang, S.W.; O’Neill, B.P.; et al. The role of factor inhibiting HIF (FIH-1) in inhibiting HIF-1 transcriptional activity in glioblastoma multiforme. PLoS ONE 2014, 9, e86102.
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56.
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512, 157–162.
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Semenza, G.L.; Van Obberghen, E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 2002, 277, 27975–27981.
- Frede, S.; Stockmann, C.; Freitag, P.; Fandrey, J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem. J 2006, 396, 517–527.
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J. Biol. Chem. 2006, 281, 33095–33106.
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci. 2018, 109, 560–571.
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacety-lating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878.

