 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Valeria Conti | + 3311 word(s) | 3311 | 2021-01-28 04:34:31 | | | |
| 2 | Peter Tang | Meta information modification | 3311 | 2021-02-17 15:58:21 | | |
Video Upload Options
Rheumatoid arthritis (RA) is a chronic inflammatory disease that is very complex and heterogeneous. If not adequately treated, RA patients are likely to manifest excess of morbidity and disability with an important impact on the quality of life. Pharmacological treatment is based on the administration of the disease-modifying antirheumatic drugs (DMARDs), subdivided into conventional synthetic (csDMARDs), targeted synthetic (tsDMARDs), and biological (bDMARDs). bDMARDs are now frequently administered in patients, both as alternative treatment and together with csDMARDs. Unfortunately, there is a therapeutic response variability both to old and new drugs.
1. Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory, autoimmune disorder affecting nearly 1% of the general population [1]. Women suffer from RA three times more often than men. Indeed, the prevalence of immune-mediated inflammatory diseases is lower in male subjects, and male RA patients appear to react better to specific treatments, such as anti-cytokines and antiproliferative drugs [2][3]. Possible reasons could be mainly related to a greater immune response in females than in males. Moreover, lymphocytes/monocytes from female subjects show higher immune/inflammatory reactivity, suggesting involvement of sex hormones in the regulation of the immune/inflammatory response in immune-mediated diseases [4][5]. In fact, sex hormones play an important role in regulating the immune/inflammatory response in RA [6].
RA initially affects small joints, progresses to larger joints and internal organs, ultimately causing an excess of morbidity and disability with an important impact on the quality of life (QoL).
The therapy is based on a treat-to-target approach, which requires tight monitoring of the disease activity in order to implement a prompt correction of the therapy regimen. The ideal therapeutic target coincides with the clinical remission or, at the very least, the low activity disease (LDA) [1].
According to the updated recommendations for the management of RA of the European League Against Rheumatism (EULAR) [7], pharmacological therapy, based on the administration of disease-modifying antirheumatic drugs (DMARDs), should be started as soon as the diagnosis of RA is made. DMARDs may slow down or even arrest the progression of joint destruction, seeking to get the treatment target, mainly assessed by the Disease Activity Score based on 28 joint counts (DAS28) [7].
DMARDs are classified in conventional synthetic (csDMARDs), targeted synthetic (tsDMARDs), and biological (bDMARDs). All the DMARDs are indicated for both non-rheumatic and rheumatic diseases thanks to their various activities, mainly immunomodulatory and anti-inflammatory ones.
Among csDMARDs, methotrexate (MTX) represents the mainstay of RA treatment.
MTX is an antimetabolite that interferes with the folate pathway binding to dihydrofolate reductase (DHFR) and thus inhibiting the transformation of dihydrofolic acid (FH2) into tetrahydrofolic acid (FH4), which is an important cofactor for the synthesis of nucleic acids and amino acids [8][9].
Under the last recommendation of the European Medicine Agency (EMA), MTX must be administered only once a week, starting with a low dose and then performing a titration as needed [10]. Serious side effects, including liver problems, bone marrow depression, and even death, occur when MTX is taken more often [10] The maximum weekly dose varies depending on the patient’s ethnic group, considering variables such as body weight and genetic factors [1].
Besides MTX, other csDMARDs suitable for RA are leflunomide and sulfasalazine. These are used as part of the first therapeutic strategy in patients with a contraindication or who manifest an early intolerance to MTX [1].
Today, besides the conventional options, clinicians have other therapeutic options, represented by ts- and bDMARDs. Tofacitinib, baricitinib, filgotinib, and upadacitinib are tsDMARDs that work by inhibiting the activity of one or more enzymes belonging to the Janus kinase family. The EULAR has included tsDMARDs among the drugs to administer in second- and later-line treatment with or without MTX. They are used in patients with moderate to severe RA who are non-responders or intolerant to csDMARDs [1].
Biological DMARDs include infliximab, adalimumab, etanercept, golimumab, and certolizumab pegol, which are all anti-tumor necrosis factor α (TNFα), and abatacept, rituximab, tocilizumab, and sarilumab. According to the EULAR recommendations [1], these drugs must be started in second- and third-line therapy, with or without MTX.
It is useful to combine pharmacological and non-pharmacological treatments with the promotion of a healthy and active lifestyle to reach the treatment target. However, despite huge advances in clinical management, diagnostic tool, and pharmacological therapy, not all patients reach an adequate response.
A variable response occurs, in fact, both to drugs and nonpharmacological therapy; therefore, there is a need to deeply investigate the factors involved in such variability observed in daily clinical practice [1].
2. Role of Autoantibodies in Predicting the Response to RA Treatment
In RA, many antibodies, such as anti-citrullinated protein antibodies (ACPAs), rheumatoid factor (RF), and anti-carbamylated protein (anti-CarP), with diagnostic and prognostic role have been identified. All of these are principally present in RA patients’ serum and synovial fluid (SF), but ACPAs and RF are also found in lung, sputum, periodontium, intestine, and cervical–vaginal mucosa [11]. For this reason, RF and ACPAs are routinely used, mainly as biomarkers for diagnostic purpose [12]. Indeed, the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) has also introduced RF and ACPAs in the diagnostic criteria of RA, and it suggests their use in guiding the choice of treatments to prevent the development of symptomatic RA [13]. The ACPAs are the most relevant diagnostic and prognostic biomarkers for RA [14] because their determination permits to classify RA patients into seropositive versus seronegative, as well as RF measurement. The ACPAs include a group of autoantibodies directed against peptides and proteins that are present in 70–90% of RA patients and have high disease-specificity (90–95%) [15]. In fact, unlike the RF, they are rarely found in other diseases or healthy subjects. In a meta-analysis by Nishimura et al., ACPAs showed a sensitivity similar to RF (≈67% vs. ≈69%), but a higher specificity (≈95% vs. ≈85%) [16]. These autoantibodies are predictive for RA development in patients without a previous diagnosis, and for more severe disease [17]. The autoimmune response in RA is presumably initiated by citrullination of self-peptides, leading to alterations of their properties. ACPAs are locally produced in RA joints, where proteins are citrullinated during the inflammatory process [18]. Citrullination [19] leads to the activation of complex immune responses and specific ACPA generation, found in approximately 75% of RA patients [20]. The risk of developing RA also depends on hereditary factors [21], and over 100 genetic loci are involved in an increased risk of RA [22]. Interestingly, twin studies have found no difference in heritability in subsets of ACPA-positive and ACPA-negative RA [23]. Instead, an analysis of large Swedish population registers showed that inheritability accounted for around 50% of ACPA-positive RA, but only 20% of ACPA-negative RA [24]. In several studies, HLA-DRB1 alleles [25], including DRB1*0401, *0404/*0408, *0405, *0101, *1001, and *1402, presenting a similar sequence at amino acid positions 67–74 in the third hypervariable region of the DR molecule (called the “shared epitope”) were associated with RA. Similarly, susceptibility to RA is associated with certain DQB1/DQA1 combinations (DQB1*03 and *04 combined with DQA1*03 (referred to as DQ3), and DQB1*0501 combined with DQA1*0101), while RA protection is mediated by other DRB1 alleles (*0103, *0402, *1102, *1103, *1301, *1302, and *1304) [26]. In North American white people, the DRB1*0401 and *0404 alleles are the most prevalent, and these genes are associated with the highest risk of severe disease. In a recent study, Wysocki et al. underlined that HLA-DRB1 genotyping is neither used in clinical practice, nor included in the current RA classification criteria [20]. Nonetheless, some HLA-DRB1 variants seem to predict the unfavorable course of the disease, including a higher risk of radiographic damage progression, higher incidence of interstitial lung disease, or lymphoproliferative diseases. The authors also suggested the possible identification of high-risk patients bearing the HLA-DRB1 risk allele, with a consequent important role of this marker in the personalization of the therapy [20]. Analyzing data from patients with early RA treated according to different strategies, by considering DQB1 typing and DRB1 typing and subtyping, Lard et al. showed that the association between the HLA class II antigens and joint destruction is affected by early and aggressive therapy with DMARDs. This interaction was independent of other prognostic factors, such as RF and baseline disease activity. Then, the authors suggested that an early and aggressive treatment with DMARDs, such as MTX, sulfasalazine, and cloroquine, can regulate the immune process, maybe by preventing a secondary release of autoantigens, by modulating the autoantigen presentation by APCs to the CD4+ T cells, and/or by inhibiting the response of T cells [27]. RFs, first identified autoantibodies in RA, are also present in infectious diseases, wherein they facilitate the removal of pathogens, and in cancer [28][29]. RFs and ACPAs show a broad spectrum of isotypes (immunoglobulin (Ig)M, IgG, IgA) [30]. The RF is an autoantibody that recognizes domains CH2 and CH3 of the Fc portion of human immunoglobulin G (IgG). According to different studies, RF testing in RA patients has a sensitivity that varies from 60 to 90% and specificity between 48 and 92% [31]. The limited specificity of RF is due to its presence also in healthy controls and patients with other autoimmune and non-autoimmune diseases [32]; consequently, RF alone is insufficient for diagnosis. High RF levels are related to an increased risk of developing RA, which may increase 26 times if they are >100 IU/mL [33], while the presence of the IgA isotype is associated with extra-articular manifestations [34]. Due to RF variable specificity, ACPA testing has been added in the latest RA classification criteria in order to increase the specificity. In RA, 50–70% of patients are positive for ACPAs, which recognize several citrullinated antigens, such as α-enolase, fibrinogen, filaggrin, vimentin, and type II collagen [35][36]. In various diseases, IgG and IgA avidity is associated with different clinical response and, in the case of ACPAs, low avidity IgG is linked to more severe bone damage [37]. About 50% of RA patients have an abnormal serologic profile several years before the development of symptoms. Moreover, elevated serum level of IgM-RF or ACPAs in healthy subjects indicates a high risk for RA onset [38]. Anti-CarP recognize other post-translational modified (PTM)-antigens and often occur together with other antibodies (e.g., ACPAs) with which they can cross-react. Anti-CarP are prognostic of the new RA onset, being detectable years before, and increasing shortly before the RA onset [39], and are predictive of joint destruction, especially in ACPA-negative RA patients [40], and they represent an important serological marker in ACPA-negative RA patients because predictive of more severe disease, as shown by radiological progression [40].
In antibody-positive patients, an increase of IgA plasmablast in peripheral blood is identified, suggesting that mucosal immune responses are involved in early RA pathogenesis [41], as well as the presence of ACPAs and RFs in the subclinical stage [42]. Therefore, Anti-CarPs, ACPAs, and RFs have a predictive role, appearing 1–3 years before RA symptoms onset, and their presence is associated with joint destruction, contributing into RA pathogenesis and development [43]. ACPAs and RFs could enhance the secretion of neutrophil extracellular traps (NETs), which show a pro-inflammatory effect, exposing autoantigens [44]. In RA patients, ACPAs are associated with parenchymal lung abnormalities [45] and may have a role in bone erosion affecting osteoclasts and increasing osteoclast precursor differentiation in vitro and in vivo [46]. This is very important because RA is characterized by chronic inflammation and bone erosion, also caused by immune complexes (ICs) composed of autoantibodies that worsen pro-inflammatory status, leading to complement activation and increased levels of chemokines and cytokines [47][48][49][50]. In a cohort of 200 RA patients, it has been demonstrated that RF testing, followed by ACPA and anti-RA33 analysis, can be a useful workflow in diagnosing the early inflammatory joint damage [51].
3. Pharmacogenetic Biomarkers
Up until now, there are no data on the role of genetic polymorphism concerning tsDMARDs. On the contrary, several studies highlighted the impact of the pharmacogenetics (PGx) to personalize the therapy with cs- and bDMARDs in patients suffering from RA.
3.1. Pharmacogenetics of csDMARDs
Owing to its efficacy, long-term safety, dose-titratable range, and cost-effectiveness, methotrexate (MTX) is still one of the most important drugs used in RA therapy.
MTX is recommend as first-line treatment in patients with RA, both as monotherapy and as an “anchor drug” in association with other DMARDs [7][52]. Following oral administration, it is absorbed from the small intestine via folate transporters and then converted through hepatic and intracellular metabolism into polyglutamated forms (MTX-PGs). Within the cell, MTX-PGs bind to and inhibit dihydrofolate reductase (DHFR) and other folate pathway enzymes required for purine and pyrimidine synthesis [53].
Despite its widespread use, the mechanism by which MTX exerts its therapeutic effects in RA is not fully understood. The reason is that MTX has anti-inflammatory effects, mediated through a variety of molecular pathways that do not necessarily involve the folate antagonism. Multiple mechanisms appear to be involved, including increase and release of adenosine; inhibition of methyl-donor production; generation of radical oxygen species (ROS); downregulation of adhesion molecules levels, eicosanoids, and matrix metalloproteinases; and interference with T cell activity and cytokine secretion [53].
Unfortunately a considerable number of patients with RA (nearly 30–40%) do not respond to MTX adequately, and many of them discontinue the therapy often because of MTX-related adverse drug reactions (ADRs). The most frequently reported ADRs include ulcerative stomatitis, leukopenia, nausea, and abdominal distress but also malaise, undue fatigue, chills and fever, dizziness, and infections [54].
However, at present, there are no biomarkers or models to predict MTX responsiveness so robust as to effectively tailor the MTX treatment in patients with RA.
Many investigations, using various technical approaches, have been proposed with the main aim to stratify the patients on their risk to not respond to MTX. For example, mRNA expression profiling [55][56][57] are considered biomarkers valuable to predict patients’ clinical outcomes before the beginning of the treatment.
In this context, the MTX PGx may be very helpful. Actually, it is well known that, besides disease-specific characteristics, numerous factors, including drug–drug interaction and patient’s genetic background may influence both the MTX efficacy and tolerability [58][59].
3.2. Pharmacogenetics of Other csDMARDS
Leflunomide (LEF) is used in patients with a contraindication to MTX. It is a prodrug that is converted into the active form A77 126 (teriflunomide). This form inhibits the dihydroorotate dehydrogenase (DHODH), an enzyme involved in the biosynthesis of pyrimidines. More than 50% of patients fail to respond to this treatment because of genetic polymorphisms.
It has been reported that the efficacy of the therapy with LEF is superior in men in comparison with women. In fact, estrogens may contrast the anti-inflammatory action of leflunomide by enhancing cytokines production.
Figure 1 reports the main pharmacogenetic biomarkers associated with csDMARD treatment.
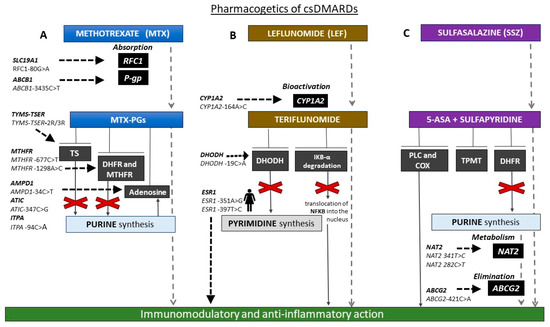
Figure 1. Schematic representation of the main pharmacogenetic biomarkers associated with conventional synthetic disease-modifying antirheumatic drug (csDMARD) treatment. (A) Methetrexate (MTX) is absorbed by reduced folate carrier 1 (RFC1) transporter, encoded by human solute carrier family 19 member 1 (SLC19A1). Patients with RFC1-80AA genotype responded better to MTX than -80AG and -80GG individuals. ATP-binding cassette subfamily B member 1 (ABCB1)-3435CT single nucleotide polymorphism (SNP) may influence the MTX absorption and therapy response. MTX is converted into MTX-polyglutamates (MTX-PGs), which in turn inhibit folate pathway enzymes thymidilate synthase (TS), dihydrofolate reductase (DHFR), and methylene tetrahydrofolate reductase (MTHFR) and increase adenosine release. Individuals carrying -3R allele of the genetic variant in the enhancer region of thymidylate synthase (TYMS; thymidylate synthase enhancer region (TSER)), encoding TS (TYMS-TSER-2R/3R) have higher TYMS mRNA expression than those with -2R and require a higher MTX dosage. MTHFR -677CT and -A1298AC SNPs lead to lower MTHFR enzyme activity. Moreover, MTX response and toxicity may be affected by SNPs in genes involved in adenosine signaling (adenosine monophosphate deaminase 1 (AMPD1)-34C>T, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase (ATIC)-347C>G, inosine triphosphate pyrophosphatase (ITPA)-94C>A). (B) Leflunomide (LEF) is converted into the active form, teriflunomide, mainly by CYP1A2. CC genotype of CYP1A2-164AC SNP is associated with increased LEF toxicity. Teriflunomide inhibits dihydroorotate dehydrogenase (DHODH; enzyme involved in the biosynthesis of pyrimidines) and nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha (IκBα) degradation. DHODH-19CC patients respond better than -19AA. The efficacy of LEF is superior in patients with ESR1 rs9340799 AA and ESR1 rs2234693 TT genotypes. (C) Polymorphisms in genes coding for N-acetyltransferase 2 (NAT2) and ATP-binding cassette subfamily G member 2 (ABCG2), involved, respectively, in drug metabolism and elimination, could influence response and safety of sulfasalazine. Patients carrying loss of function (LoF) allele A of the ABCG2 -421CA SNP respond better than carriers of LoF allele of NAT2-341TC, and -282CT is associated with high-risk toxicity. csDMARDs, conventional synthetic disease-modifying antirheumatic drugs; MTX, methetrexate; SLC19A1, human solute carrier family 19 member 1; RFC1, reduced folate carrier 1; P-gp, P-glycoprotein; ABCB1, ATP-binding cassette subfamily B member 1; MTX-PGs, MTX-polyglutamate: TYMS-TSER, thymidylate synthase-thymidylate synthase enhancer region; TS, thymidilate synthase; DHFR, dihydrofolate reductase; MTHFR, methylene tetrahydrofolate reductase; AMPD1, adenosine monophosphate deaminase 1; ATIC, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase; ITPA, inosine triphosphate pyrophosphatase; DHODH, dihydroorotate dehydrogenase; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; ESR1, estrogen receptor 1; TPMT, thiopurine methyltransferase; 5-ASA, 5-aminosalicylic acid; TPMT, thiopurine S-methyltransferase; NAT2, N-acetyltransferase 2; ABCG2, ATP-binding cassette subfamily G member 2.
3.3. Pharmacogenetics of bDMARDs
Biological DMARDs are now frequently administered in patients with RA both as alternative treatment and together with csDMARDs. However, accumulating evidence demonstrates therapeutic response variability also for this class of drugs. Therefore, given the variety of bDMARDs now available and their high costs, the identification of pre-therapeutic predictors of response is becoming a priority.
It has been reported that 25–30% of patients with RA do not respond adequately to TNFα inhibitors [60].
The anti-TNFα PGx is not yet used in daily clinical practice. Indeed, the results are promising but yet inconclusive.
Several studies have shown that the presence of polymorphisms in the genes coding for TNFα and TNF receptor (TNFR) could influence the response to treatment. Among them, three studies evaluated the association between TNFα 308A>G (rs1800629) SNP and clinical response to infliximab [61], etanercept [62], and adalimumab [63], independently. All of these studies showed that RA patients with a TNFα-308 G/G genotype responded to biological drugs better than patients with -308 A/G or -A/A genotypes, suggesting that TNFα-308A>G genotyping may be a useful tool for predicting the response to anti-TNF agents. Similar results were obtained in the meta-analysis [64] by Zeng et al. that, including 15 studies with a total of 2127 RA patients, suggests that TNFα-308G allele plays a major role in guiding the response to anti-TNFα treatment (OR = 1.87, 95 % CI = 1.26–2.79) [64].
Figure 2 reports the main pharmacogenetic biomarkers associated with bDMARDs treatment.
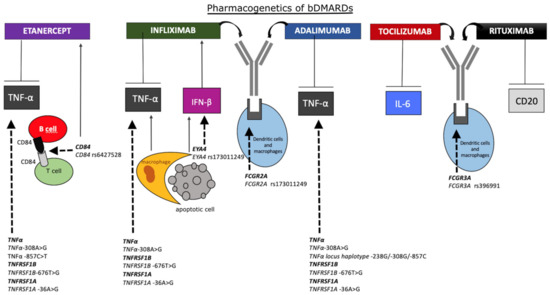
Figure 2. Schematic representation of the main pharmacogenetic biomarkers associated with bDMARDs treatment. Rheumatoid arthritis (RA) patients with tumor necrosis factor α (TNFα)-308 G/G genotype may respond to anti-TNFα drugs better than patients with -308 A/G or -A/A genotypes. TNFα -857C>T and CD84 rs6427528 SNPs also affect the treatment with etanercept. TNFα-308AA genotype is associated with a poorer response compared with TNFα-308GG, and CD84 rs6427528 SNP is positively correlated with disease activity in patients treated with etanercept. In apoptosis, DNA from dead cells is digested by DNase II in the macrophages after they are engulfed. Interferon β (IFN-β) and TNFα are produced from the macrophages carrying undigested DNA. Eyes absent homolog 4 (EYA4), originally identified as a co-transcription factor, stimulates the expression of IFN-β in response to undigested DNA of apoptotic cells, worsening the response to infliximab. The SNP rs173011249 in EYA4 gene is associated with improved therapy response. Treatment with infliximab and adalimumab may be affected by fragment crystallizable region of immunoglobulin (Ig)G receptor 2A (FCGR2A) rs173011249 in gene coding for immunoglobulin gamma Fc region receptor II-a. Patients bearing FCGR3A rs396991-TT genotype, probably lowering plasmatic clearance, respond better to tocilizumab. Rituximab is more efficacious in patients carrying FCGR3A rs396991-G allele, associated with higher affinity of FCR for the Fc region. bDMARDs, biologics disease-modifying antirheumatic drugs; TNFα, tumor necrosis factor α; TNFRSF-1B/1A, TNF receptor superfamily member 1B/1A; IFN-β, interferon β; EYA4, eyes absent homolog 4; FCGR-2A/3A, fragment crystallizable region of IgG receptor 2A/3A; IL-6, interleukin-6.
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038.
- Sokka, T.; Toloza, S.; Cutolo, M.; Kautiainen, H.; Makinen, H.; Gogus, F.; Skakic, V.; Badsha, H.; Peets, T.; Baranauskaite, A.; et al. QUEST-RA Group. Women, men, and rheumatoid arthritis: Analyses of disease activity, disease characteristics, and treatments in the QUEST-RA study. Arthritis Res. Ther. 2009, 11, R7.
- Kvien, T.K.; Uhlig, T.; Ødegård, S.; Heiberg, M.S. Epidemiological aspects of rheumatoid arthritis: The sex ratio. Ann. N. Y. Acad. Sci. 2006, 69, 212–222.
- Kanda, N.; Tsuchida, T.; Tamaki, K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 328–337.
- Kramer, P.R.; Kramer, S.F.; Guan, G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004, 50, 1967–1975.
- Cutolo, M.; Capellino, S.; Montagna, P.; Villaggio, B.; Sulli, A.; Seriolo, B.; Straub, R.H. New roles for estrogens in rheumatoid arthritis. Clin. Exp. Rheumatol. 2003, 21, 687–690.
- Smolen, J.S.; Landewé, R.; Bijlsma, J.W.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; Van Vollenhoven, R.F.; De Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 76, 685–699.
- Cronstein, B.N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172.
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023.
- Italian Medicines Agency Home Page. Available online: https://www.aifa.gov.it/documents/20142/847374/Methotrexate_public_health_communication_IT.pdf/a8fb75db-4b62-2783-528b-6701ce5cfc66 (accessed on 15 November 2020).
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid Arthritis and the Mucosal Origins Hypothesis: Protection Turns to Destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557.
- Trouw, L.A.; Rispens, T.; Toes, R.E.M. Beyond Citrullination: Other Post-Translational Protein Modifications in Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2017, 13, 331–339.
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid Arthritis Classification Criteria: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588.
- van Boekel, M.A.M.; Vossenaar, E.R.; van den Hoogen, F.H.J.; van Venrooij, W.J. Autoantibody Systems in Rheumatoid Arthritis: Specificity, Sensitivity and Diagnostic Value. Arthritis Res. 2002, 4, 87–93.
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The Diagnostic Properties of Rheumatoid Arthritis Antibodies Recognizing a Cyclic Citrullinated Peptide. Arthritis Rheum. 2000, 43, 155–163.
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-Analysis: Diagnostic Accuracy of Anti-Cyclic Citrullinated Peptide Antibody and Rheumatoid Factor for Rheumatoid Arthritis. Ann. Intern. Med. 2007, 146, 797–808.
- Verpoort, K.N.; Jol-van der Zijde, C.M.; Papendrecht-van der Voort, E.A.M.; Ioan-Facsinay, A.; Drijfhout, J.W.; van Tol, M.J.D.; Breedveld, F.C.; Huizinga, T.W.J.; Toes, R.E.M. Isotype Distribution of Anti-Cyclic Citrullinated Peptide Antibodies in Undifferentiated Arthritis and Rheumatoid Arthritis Reflects an Ongoing Immune Response. Arthritis Rheum. 2006, 54, 3799–3808.
- Reparon-Schuijt, C.C.; van Esch, W.J.; van Kooten, C.; Schellekens, G.A.; de Jong, B.A.; van Venrooij, W.J.; Breedveld, F.C.; Verweij, C.L. Secretion of Anti-Citrulline-Containing Peptide Antibody by B Lymphocytes in Rheumatoid Arthritis. Arthritis Rheum. 2001, 44, 41–47.
- Wang, S.; Wang, Y. Peptidylarginine Deiminases in Citrullination, Gene Regulation, Health and Pathogenesis. Biochim. Biophys. Acta 2013, 1829, 1126–1135.
- Wysocki, T.; Olesińska, M.; Paradowska-Gorycka, A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis-From Research to Clinical Practice. Cells 2020, 9, 1127.
- Gregersen, P.K. Genetics of Rheumatoid Arthritis: Confronting Complexity. Arthritis Res. 1999, 1, 37–44.
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery. Nature 2014, 506, 376–381.
- van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.M.; Huizinga, T.W.J.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.M.; de Vries, R.R.P. Quantitative Heritability of Anti-Citrullinated Protein Antibody-Positive and Anti-Citrullinated Protein Antibody-Negative Rheumatoid Arthritis. Arthritis Rheum. 2009, 60, 916–923.
- Frisell, T.; Holmqvist, M.; Källberg, H.; Klareskog, L.; Alfredsson, L.; Askling, J. Familial Risks and Heritability of Rheumatoid Arthritis: Role of Rheumatoid Factor/Anti-Citrullinated Protein Antibody Status, Number and Type of Affected Relatives, Sex, and Age. Arthritis Rheum. 2013, 65, 2773–2782.
- O’Dell, J.R.; Nepom, B.S.; Haire, C.; Gersuk, V.H.; Gaur, L.; Moore, G.F.; Drymalski, W.; Palmer, W.; Eckhoff, P.J.; Klassen, L.W.; et al. HLA-DRB1 Typing in Rheumatoid Arthritis: Predicting Response to Specific Treatments. Ann. Rheum. Dis. 1998, 57, 209–213.
- Zanelli, E.; Breedveld, F.C.; de Vries, R.R. HLA Class II Association with Rheumatoid Arthritis: Facts and Interpretations. Hum. Immunol. 2000, 61, 1254–1261.
- Lard, L.R.; Boers, M.; Verhoeven, A.; Vos, K.; Visser, H.; Hazes, J.M.W.; Zwinderman, A.H.; Schreuder, G.M.T.; Breedveld, F.C.; De Vries, R.R.P.; et al. Early and Aggressive Treatment of Rheumatoid Arthritis Patients Affects the Association of HLA Class II Antigens with Progression of Joint Damage. Arthritis Rheum. 2002, 46, 899–905.
- Van Snick, J.L.; Van Roost, E.; Markowetz, B.; Cambiaso, C.L.; Masson, P.L. Enhancement by IgM Rheumatoid Factor of in Vitro Ingestion by Macrophages and in Vivo Clearance of Aggregated IgG or Antigen-Antibody Complexes. Eur. J. Immunol. 1978, 8, 279–285.
- Hogben, D.N.; Devey, M.E. Studies on Rheumatoid Factor: I. The Effect of Rheumatoid Factor on the Clearance of Preformed Immune Complexes in Mice. Clin. Exp. Immunol. 1986, 66, 648–653.
- Falkenburg, W.J.J.; van Schaardenburg, D.; Ooijevaar-de Heer, P.; Wolbink, G.; Rispens, T. IgG Subclass Specificity Discriminates Restricted IgM Rheumatoid Factor Responses From More Mature Anti-Citrullinated Protein Antibody-Associated or Isotype-Switched IgA Responses. Arthritis Rheumatol. 2015, 67, 3124–3134.
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid Factors: Clinical Applications. Dis. Mark. 2013, 35, 727–734.
- Verheul, M.K.; Fearon, U.; Trouw, L.A.; Veale, D.J. Biomarkers for Rheumatoid and Psoriatic Arthritis. Clin. Immunol. 2015, 161, 2–10.
- Nielsen, S.F.; Bojesen, S.E.; Schnohr, P.; Nordestgaard, B.G. Elevated Rheumatoid Factor and Long Term Risk of Rheumatoid Arthritis: A Prospective Cohort Study. BMJ 2012, 345, e5244.
- Jónsson, T.; Arinbjarnarson, S.; Thorsteinsson, J.; Steinsson, K.; Geirsson, A.J.; Jónsson, H.; Valdimarsson, H. Raised IgA Rheumatoid Factor (RF) but Not IgM RF or IgG RF Is Associated with Extra-Articular Manifestations in Rheumatoid Arthritis. Scand. J. Rheumatol. 1995, 24, 372–375.
- Takizawa, Y.; Suzuki, A.; Sawada, T.; Ohsaka, M.; Inoue, T.; Yamada, R.; Yamamoto, K. Citrullinated Fibrinogen Detected as a Soluble Citrullinated Autoantigen in Rheumatoid Arthritis Synovial Fluids. Ann. Rheum. Dis. 2006, 65, 1013–1020.
- Burkhardt, H.; Koller, T.; Engström, A.; Nandakumar, K.S.; Turnay, J.; Kraetsch, H.G.; Kalden, J.R.; Holmdahl, R. Epitope-Specific Recognition of Type II Collagen by Rheumatoid Arthritis Antibodies Is Shared with Recognition by Antibodies That Are Arthritogenic in Collagen-Induced Arthritis in the Mouse. Arthritis Rheum. 2002, 46, 2339–2348.
- Fialová, L. Avidity of Selected Autoantibodies—Usefulness of Their Determination for Clinical Purposes. Epidemiol. Mikrobiol. Imunol. Cas. Spol. Pro Epidemiol. Mikrobiol. Ces. Lek. Spol. J.E. Purkyne 2016, 65, 155–163.
- Nielen, M.M.J.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.M.T.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A.C. Specific Autoantibodies Precede the Symptoms of Rheumatoid Arthritis: A Study of Serial Measurements in Blood Donors. Arthritis Rheum. 2004, 50, 380–386.
- Gan, R.W.; Trouw, L.A.; Shi, J.; Toes, R.E.M.; Huizinga, T.W.J.; Demoruelle, M.K.; Kolfenbach, J.R.; Zerbe, G.O.; Deane, K.D.; Edison, J.D.; et al. Anti-Carbamylated Protein Antibodies Are Present Prior to Rheumatoid Arthritis and Are Associated with Its Future Diagnosis. J. Rheumatol. 2015, 42, 572–579.
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.C.; van Veelen, P.A.; Levarht, N.E.W.; van der Helm-van Mil, A.H.M.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377.
- Kinslow, J.D.; Blum, L.K.; Deane, K.D.; Demoruelle, M.K.; Okamoto, Y.; Parish, M.C.; Kongpachith, S.; Lahey, L.J.; Norris, J.M.; Robinson, W.H.; et al. Elevated IgA Plasmablast Levels in Subjects at Risk of Developing Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 2372–2383.
- van de Stadt, L.A.; Witte, B.I.; Bos, W.H.; van Schaardenburg, D.A. Prediction Rule for the Development of Arthritis in Seropositive Arthralgia Patients. Ann. Rheum. Dis. 2013, 72, 1920–1926.
- Verheul, M.K.; Böhringer, S.; van Delft, M.A.M.; Jones, J.D.; Rigby, W.F.C.; Gan, R.W.; Holers, V.M.; Edison, J.D.; Deane, K.D.; Janssen, K.M.J.; et al. Triple Positivity for Anti-Citrullinated Protein Autoantibodies, Rheumatoid Factor, and Anti-Carbamylated Protein Antibodies Conferring High Specificity for Rheumatoid Arthritis: Implications for Very Early Identification of At-Risk Individuals. Arthritis Rheumatol. 2018, 70, 1721–1731.
- Husby, G.; Gran, J.T.; Johannessen, A. Epidemiological and Genetic Aspects of IgM Rheumatoid Factors. Scand. J. Rheumatol. Suppl. 1988, 75, 213–218.
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engström, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural Changes and Antibody Enrichment in the Lungs Are Early Features of Anti-Citrullinated Protein Antibody-Positive Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 31–39.
- Roosnek, E.; Lanzavecchia, A. Efficient and Selective Presentation of Antigen-Antibody Complexes by Rheumatoid Factor B Cells. J. Exp. Med. 1991, 173, 487–489.
- Daha, N.A.; Banda, N.K.; Roos, A.; Beurskens, F.J.; Bakker, J.M.; Daha, M.R.; Trouw, L.A. Complement Activation by (Auto-) Antibodies. Mol. Immunol. 2011, 48, 1656–1665.
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.N.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.J.; Toes, R.E. Anti-Cyclic Citrullinated Peptide Antibodies from Rheumatoid Arthritis Patients Activate Complement via Both the Classical and Alternative Pathways. Arthritis Rheum. 2009, 60, 1923–1931.
- Aleyd, E.; Al, M.; Tuk, C.W.; van der Laken, C.J.; van Egmond, M. IgA Complexes in Plasma and Synovial Fluid of Patients with Rheumatoid Arthritis Induce Neutrophil Extracellular Traps via FcαRI. J. Immunol. 2016, 197, 4552–4559.
- Paoliello-Paschoalato, A.B.; Marchi, L.F.; de Andrade, M.F.; Kabeya, L.M.; Donadi, E.A.; Lucisano-Valim, Y.M. Fcγ and Complement Receptors and Complement Proteins in Neutrophil Activation in Rheumatoid Arthritis: Contribution to Pathogenesis and Progression and Modulation by Natural Products. Evid. Based Complement. Altern. Med. 2015, 2015, 429878.
- Nell, V.P.K.; Machold, K.P.; Stamm, T.A.; Eberl, G.; Heinzl, H.; Uffmann, M.; Smolen, J.S.; Steiner, G. Autoantibody Profiling as Early Diagnostic and Prognostic Tool for Rheumatoid Arthritis. Ann. Rheum. Dis. 2005, 64, 1731–1736.
- Singh, J.A.; Saag, K.G.; Bridges, S.L.J.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2016, 68, 1–25.
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine 2019, 86, 301–307.
- Food and Drug Administration Home Page. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/008085s068lbl.pdf (accessed on 15 November 2020).
- Plant, D.; Maciejewski, M.; Smith, S.; Nair, N.; Hyrich, K.; Ziemek, D.; Barton, A.; Verstappen, S. Profiling of Gene Expression Biomarkers as a Classifier of Methotrexate Nonresponse in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 678–684.
- Singh, A.; Patro, P.S.; Aggarwal, A. MicroRNA-132, MiR-146a, and MiR-155 as Potential Biomarkers of Methotrexate Response in Patients with Rheumatoid Arthritis. Clin. Rheumatol. 2019, 38, 877–884.
- Sode, J.; Krintel, S.B.; Carlsen, A.L.; Hetland, M.L.; Johansen, J.S.; Hørslev-Petersen, K.; Stengaard-Pedersen, K.; Ellingsen, T.; Burton, M.; Junker, P.; et al. Plasma MicroRNA Profiles in Patients with Early Rheumatoid Arthritis Responding to Adalimumab plus Methotrexate vs Methotrexate Alone: A Placebo-Controlled Clinical Trial. J. Rheumatol. 2018, 45, 53–61.
- Mikkelsen, T.S.; Thorn, C.F.; Yang, J.J.; Ulrich, C.M.; French, D.; Zaza, G.; Dunnenberger, H.M.; Marsh, S.; McLeod, H.L.; Giacomini, K.; et al. PharmGKB Summary: Methotrexate Pathway. Pharmacogenet. Genom. 2011, 21, 679–686.
- Qiu, Q.; Huang, J.; Lin, Y.; Shu, X.; Fan, H.; Tu, Z.; Zhou, Y.; Xiao, C. Polymorphisms and Pharmacogenomics for the Toxicity of Methotrexate Monotherapy in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Medicine 2017, 96, e6337.
- Salliot, C.; Finckh, A.; Katchamart, W.; Lu, Y.; Sun, Y.; Bombardier, C.; Keystone, E. Indirect Comparisons of the Efficacy of Biological Antirheumatic Agents in Rheumatoid Arthritis in Patients with an Inadequate Response to Conventional Disease-Modifying Antirheumatic Drugs or to an Anti-Tumour Necrosis Factor Agent: A Meta-Analysis. Ann. Rheum. Dis. 2011, 70, 266–271.
- Mugnier, B.; Balandraud, N.; Darque, A.; Roudier, C.; Roudier, J.; Reviron, D. Polymorphism at position -308 of the tumor necrosis factor alpha gene influences outcome of infliximab therapy in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 1849–1852.
- Guis, S.; Balandraud, N.; Bouvenot, J.; Auger, I.; Toussirot, E.; Wendling, D.; Mattei, J.P.; Nogueira, L.; Mugnier, B.; Legeron, P. Influence of -308 A/G polymorphism in the tumor necrosis factor alpha gene on etanercept treatment in rheumatoid arthritis. Arthritis Rheum. 2007, 57, 1426–1430.
- Cuchacovich, M.; Soto, L.; Edwardes, M.; Gutierrez, M.; Llanos, C.; Pacheco, D.; Sabugo, F.; Alamo, M.; Fuentealba, C.; Villanueva, L.; et al. Tumour necrosis factor (TNF)alpha -308 G/G promoter polymorphism and TNFalpha levels correlate with a better response to adalimumab in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2006, 35, 435–440.
- Zeng, Z.; Duan, Z.; Zhang, T.; Wang, S.; Li, G.; Gao, J.; Ye, D.; Xu, S.; Xu, J.; Zhang, L. Association between tumor necrosis factor-α (TNF-α) promoter -308 G/A and response to TNF-α blockers in rheumatoid arthritis: A meta-analysis. Mod. Rheumatol. 2013, 23, 489–495.


