 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalia Komarova | + 4620 word(s) | 4620 | 2021-01-13 06:59:23 | | | |
| 2 | Camila Xu | + 93 word(s) | 4713 | 2021-02-05 08:02:37 | | |
Video Upload Options
Aptamers, short single-stranded DNA or RNA molecules capable of specific binding to a target of interest, are gaining in research interest due to their versatile application potential as analytical, diagnostic, and therapeutics agents.
1. Introduction
Aptamers offer the advantages of low synthesis cost, small size, thermal stability, ease of labeling, and ability of regeneration. aptamers are usually obtained using the systematic evolution of ligands by exponential enrichment (SELEX) process, which was first implemented in the 1990s [1][2]. During its three decades in existence, SELEX has obtained modifications aiming to extend the target range and improve the efficiency and speed of the selection process.
SELEX is an iterative procedure of selection of target-binding species from a vast library of random oligonucleotides. The random nucleic acid library is incubated with the target molecules to facilitate the formation of a complex between the target and oligonucleotides displaying target affinity. Target-bound species are then separated from the rest of the unbound library pool and PCR-amplified. The new ssDNA/RNA library enriched with target-binding oligonucleotides is then regenerated from the PCR product and involved in the novel selection cycle. The selection continues until the library is sufficiently enriched.
The progress of enrichment can be monitored with the simple detection of the amount of target-bound nucleic acid by measuring the affinity of the selected oligonucleotide pool to the target or by assessing the decrease in the diversity of the selected library. In conventional SELEX, the terminal library after the selection is cloned and 30–100 representatives are sequenced with Sanger sequencing. Few of them are picked either randomly or based on sequence analysis for further experimental evaluation of their affinity and specificity to the target, after which the candidate oligonucleotide can be nominated as the aptamer. In that respect, the correct identification of candidate aptamers is a key point for the overall selection success [3].
The huge progress in DNA sequencing technologies, enabling high-throughput sequencing (HTS) of DNA and RNA molecules of different lengths, has occurred within the past fifteen years with the establishment of next-generation sequencing technologies that found an application in SELEX technology. The starting library for aptamer selection normally contains 1011–1016 unique nucleic acid species; the library diversity is practically never sufficiently reduced with enrichment under SELEX to a size that can be effectively covered with Sanger sequencing. Therefore, the ability to identify up to millions of sequences provided by HTS instead of hundreds with classic sequencing perfectly fits the SELEX procedure.
The enlarged number of recognized SELEX-derived oligonucleotide sequences enables more precise candidate aptamer identification. In addition, HTS applied for cycle-to-cycle enriched libraries can elucidate the constraints and pitfalls of the aptamer isolation process, which in turn can provide the basis for rendering SELEX more efficiently. The main drawback of HTS is the generation of an enormous amount of data to be analyzed. The potential of HTS data is often not realized due to the complexity of data processing, the need for high expertise in bioinformatics, and the lack of easy-to-use software [4].
2. Sequencing Platforms
Next generation sequencing techniques include several different strategies that allow short or long reads of the target nucleic acid molecule [5]. High-throughput sequencing in SELEX is mainly shared between the Illumina, 454 Roche, and IonTorrent sequencing platforms. The Illumina sequencing platform is believed to be the most widespread [6]. All of these platforms employ sequencing-by-synthesis (SBS) techniques and enable short reads of the target nucleic acids. The read length is limited to 300–400 nt depending on the specific device. Commonly, the starting SELEX library does not exceed 150 nt [3], and the length of short-read sequencing is sufficient to read the entire sequence of library representatives.
In SBS technology, the sequenced DNA strand serves as the template for the synthesis of the complementary strand, and the sequencing device identifies the bases incorporated into this synthesized strand. Before sequencing, the target DNA is clonally amplified to achieve localization of the same single-stranded sequence clones at the distinct area of the sequencing chip. This cloning serves for signal amplification during the sequencing process (Figure 1). In Illumina technology (Figure 1a), the clones of each single template molecule are amplified directly above the sequencing chip surface using bridge amplification [5]. As a result, the clones of each initial template sequence form a cluster of a detectable size on the chip.
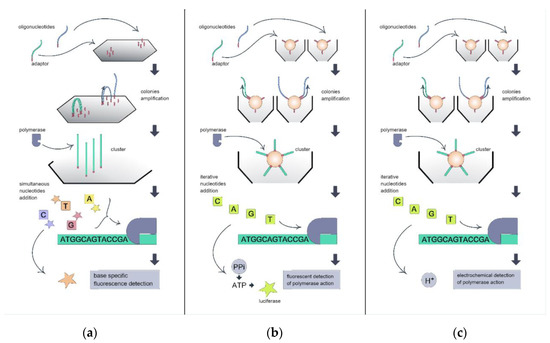
Figure 1. Illustration of sequencing-by-synthesis technology variants implied by different high-throughput sequencing platforms: (a) Illumina; (b) 454 Roche; and (c) IonTorrent.
The colonies of the ssDNA template are then hybridized with the sequencing primer. Then, the DNA polymerase begins the synthesis of the complementary strand. Four fluorescently modified dNTPs are simultaneously added to the chip surface for each synthesis step. At each colony cluster, the base-specific fluorescent signal is formed with base incorporation, and this signal is recognized by the device detector. In 454 Roche (Figure 1b) and IonTorrent (Figure 1c) devices, clones of a single template molecule are formed on the surface of the beads using the emulsion PCR process. A single bead, a single template molecule, DNA polymerase, dNTPs, and primers are captured in the emulsion drop, and, after amplification, the bead is covered with the clones of the template.
Then, the beads are distributed above the cell-structured sequencing chips, enabling further signal detection. Four different dNTPs are iteratively added to the chip. If the added dNTP is complementary to the template DNA, the dNTP incorporates the synthesized strand, and DNA polymerase action generates the signal to be detected by the device. In 454 Roche devices, the release of pyrophosphate during each DNA polymerization step is fluorescently detected with the use of an enzyme cascade. IonTorrent devices electrochemically detect the release of H+ at each DNA polymerization step using an ion-sensitive field effect transistor (ISFET).
Next generation sequencing (NGS) techniques enabling long reads such as Nanopore sequencing are less applicable for sequencing SELEX libraries. Theoretically, long reads can be useful in genomic RNA SELEX, where the initial library species can be longer than in conventional SELEX [7]. The ability of nanopore sequencing for the detection of modified nucleobases can also be applicable in the process of isolating aptamers containing modified and unnatural bases [6].
3. Sample Preparation for High-Throughput Sequencing (HTS)
HTS always requires special library preparation before sequencing. In SBS, library preparation is intended to enable the correct clonal amplification of the substrate and to introduce the universal primer binding site for further enzymatic synthesis of the complementary strand for signal generation. For this purpose, the ssDNA/RNA pool obtained from SELEX is transformed to dsDNA flanked with special adaptor sequences. These adaptors contain specific sequences that serve both the hybridization of the target DNA with the complementary short ssDNA strands on the surface of the solid support for further clonal amplification and in the priming of DNA synthesis. In addition to the regions necessary for hybridization and amplification of the template, the adaptor sequences can bear specific indices or barcodes to encode the sample, which allows for the sequencing of multiple samples within a single chip. This option is especially relevant for the high-throughput sequencing of the SELEX libraries obtained from cycle to cycle under the selection process.
Sequencing adaptors and kits for their incorporation can be purchased from commercial suppliers. The sample preparation workflow for different SBS platforms is similar and includes the following steps: the conversion of ssDNA or RNA to dsDNA, end repair (blunt-ending and the phosphorylation of 5′-ends), adenylation of 3′-ends, ligation of adapters, selection of correct ligation product, and amplification of the adaptor-ligated product (Figure 2a). The last PCR-amplification step is optional, and should be avoided for SELEX libraries to escape additional PCR bias [8][9]. This also shortens the sample preparation time.
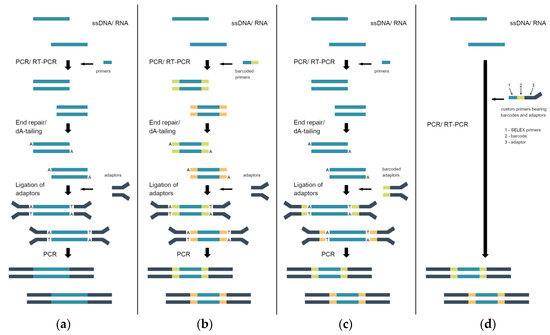
Figure 2. Sample preparation workflows for sequencing library preparation. (a) The common supplier’s workflow for the preparation of non-indexed libraries. (b) The workflow for the preparation of indexed libraries; barcodes are introduced at the first PCR amplification step with the custom primers. (c) The workflow for the preparation of indexed libraries; barcodes are introduced at the ligation step using the commercial adaptors. (d) Single PCR workflow for the preparation of indexed libraries; barcodes and adaptors are introduced with the custom primers.
The only requirement is the enlarged amount of the starting dsDNA sample, which is affordable in most SELEX experiments. The majority of sample preparation kits are developed for the preparation of sequencing libraries for large DNA and RNA molecules and, therefore, assume mechanical or enzymatic fragmentation of the sample. For SELEX-derived libraries, this step is not needed [10]. The majority of protocols for commercial kits also employ the usage of magnetic AMPure XP beads for the size-selection of DNA fragments after the adaptor ligation step. SELEX-derived samples are typically too small for bead-based separations and, therefore could be better (but not obligatory [11]) processed using gel-based separations and extraction [10]. Both agarose and polyacrylamide gels are acceptable.
As an example, a detailed sample preparation protocol for Illumina sequencing based on a commercial PCR-free adapter ligation reagent kit is provided in [10]. Interestingly, it was suggested that sample specific barcodes be introduced in the first step of ssDNA PCR conversion to dsDNA with indexed primers instead of using indexed commercial adaptors (Figure 2b). The same sample barcoding approach was used in [12]. This makes sample preparation less expensive if sample pooling is used as it enables using only the adapter sequence for the final library, and this is especially relevant when ordering a commercial sequencing service [12]. Alternatively, barcode-containing adaptors can be used if necessary (Figure 2c).
The presence of fixed flanking regions in SELEX libraries allows one to omit the ligation of commercial adapters. For example, in [13], the samples obtained from different selection cycles were PCR-amplified with custom primers bearing priming sequences specific for the SELEX library, barcode sequences for encoding the samples from different selection cycles, and Illumina adapter sequences P5/P7 (Figure 2d). The sequencing sample was generated using only one PCR-amplification step with no need for subsequent adaptor ligation and additional amplification. The same approach employing fusion primers can be applied for other sequencing platforms [11][14][15].
A special library preparation protocol for Illumina sequencing is described for genetic alphabet expansion SELEX (ExSELEX), which employs an unnatural base pair system for the initial SELEX library [16]. SBS is normally unavailable for the libraries containing modified and unnatural nucleobases. To make the library compatible with deep sequencing, a special protocol was developed [16]. The initial SELEX library is designed to contain a few unnatural bases in each representative sample. Each unnatural base position is concatenated with a specific sequence of natural bases. After the selection process, the enriched library proceeds with two-step amplification in which unnatural bases are substituted with natural ones and, thus, becomes SBS-compatible. After sequencing, the known specific sequences serve as a kind of a barcode to recognize the positions of the unnatural base.
The sequencing libraries require thorough validation before sequencing because the efficiency of clonal amplification, and thus the sequencing output, depends strongly on the library concentration. Both the library length confirming the correct insertion of the initial template between the adaptor sequences and the library concentration should be checked. The library size can be confirmed using standard gel separation or the Bioanalyzer system. The best method for the determination of the library concentration is real-time PCR with special kits for the validation of NGS libraries as this specifically detects the adaptor-containing sequences that are essential for clonal amplification [10]. However, if the library is clean enough and displays the correct size with no by-products, the quantification can be done with sensitive fluorimeters like Qubit [10]. If the multiplexed library is sequenced, the concentration of different samples should be normalized before sample pooling to achieve equal (or desired) reads distribution between the sequences.
4. Processing of HTS Data
The use of HTS-SELEX techniques for aptamer selection requires new bioinformatics approaches to analyze the huge amount of information produced by NGS that would make the identification of aptamer candidates based on the sequencing results more transparent and reasonable. Simultaneously with the development of software for direct HTS-SELEX data analysis, software solutions appeared for practical research issues related to HTS-SELEX, for instance, reducing SELEX cycles [17][18][19][20][21], determining selection conditions [22], or even searching for binding sites [23][24]. The standard protocol for the analysis of HTS results can be divided into several stages: data preprocessing, primary enrichment analysis, library clustering, and a binding motifs search. Separate bioinformatics instruments to perform each of these steps individually exist, and many of them have been further integrated into more complex software packages to perform full-scale analysis of HTS data.
Data preprocessing steps depend on the structure of the library and the sequencing method [18]; however, in general, preprocessing consists of the removal of sequencing artifacts, filtering by sequence length, replacement of reverse sequences, and, in some cases, removal or masking regions of constant sequence. Initially, each laboratory solved these problems individually using in-house developed software [19][25], and also adopted Tallymer [26] software initially developed for k-mer sequence counting by setting the k-value to the length of the library random region [17]. Researchers with advanced computer competences can easily perform these tasks, but special programs and adaptations of existing platforms also appear to manage HTS-SELEX preprocessing. However, data preprocessing scripts are still aimed toward the classic SELEX design and are not adapted to other variations. For example, many programs do not allow for the masking of constant regions that are not at the edges of sequences, much like the Capture region in oligonucleotides when a selection has been performed according to the Capture-SELEX protocol [27].
The Galaxy Project is a convenient solution for researchers without bioinformatics skills. The platform is easy to use and contains a large number of standard text and bioinformatics tools that allow all steps of raw NGS data preprocessing to be performed [28]. W. H. Thiel compiled a workflow containing all the necessary stages of basic data preprocessing and the first stages of analysis to eliminate unenriched sequences [29] so that the initial pool of sequences could be reduced by more than 90%, facilitating and accelerating subsequent analysis. Another special program developed for the preprocessing of HTS-SELEX data is AptaPLEX [30], which is described further as it was included in a large program package for full HTS-SELEX data analysis.
Primary estimation of the enrichment of SELEX libraries is based on counting the copy number of each sequence in different rounds and identifying the number of rounds in which the sequence is present. These data can serve for the evaluation of the enrichment rate of each unique sequence throughout the library evolution under the selection pressure [19]. The Galaxy server offers tools for creating a non-redundant sequences database, which can further be exported and used for further analysis of the sequence abundance scoring and their cycle-to-cycle enrichment rate using any other desired instrument. These parameters, which are also calculated by most other programs, are used to track the dynamics of selection and are the basis for the identification of aptamer candidates. However, an assumption that the most enriched sequences take the lead in selection due to their ability to bind to the target is not always confirmed empirically, particularly with the increase in selection cycles [19][25][31]. Therefore, different programs perform sequence clusterization or binding motifs in the search for a more representative aptamer identification.
Clusterization aims to find sequence families and estimate the enrichment of each cluster instead of the enrichment of individual sequences. Clusterization, according to the primary structure, is based on the assumption that a random selection of oligonucleotides with near-identical primary structure from an underrepresented library is extremely unlikely; a supposal that this similarity is caused by some divergent biases during library processing during SELEX is more expectable. This clustering approach provides an opportunity to identify primary and mutant oligonucleotide forms, reduce the diversity of an analyzing pool, and select oligonucleotides with different structures for further screening [32]. The most common representatives of obtained clusters are likely to be different in structure and could interact with the target in different ways during selection [32].
FASTAptamer [32] is perhaps one of the most common software products for HTS-SELEX data analysis, especially in the primary stages. The program has the form of an open source script set, where each script aims at solving different problems and is usable regardless of the specifics of the SELEX experiment. The program has no graphical interface and, therefore requires at least the simplest understanding of working with the command line. FASTAptamer can be used on any operating system if the PERL interpreter is preinstalled that the program is written on. At the same time, FASTAptamer scripts are available on the previously described Galaxy platform (if it is installed on the computer) and can be combined into one workflow with data preprocessing.
The obligatory first stage of the analysis is the FASTAptamer-Count script, which uses the initially preprocessed sequencing data to calculate and normalize the frequency of unique sequences occurring in the pool (e.g., to perform primary estimation of the enrichment). The results of the FASTAptamer-Count are used by other scripts for sequence clustering, comparing the enrichment in different populations, and identifying the sequences with the highest enrichment rate over several rounds of selection. The clustering script is based on the Levenshtein distance method, which can compare sequences of different lengths [32]. PATTERNITY-seq, another clustering program, works on a similar principle, but has apparently not become widespread [33].
Another approach to cluster sequences based on their primary structure is the local hashing algorithm. Programs based on this algorithm work faster with a large amount of data; however, they are not capable of comparing sequences of different lengths, and therefore all sequences must be filtered to the same length before analysis [34]. This approach has been implemented in the programs AptaCluster [35] and SEWAL [36]. AptaCluster is described further as AptaSUITE. SEWAL was adapted for the analysis of Illumina results and provides analysis results in the form of three-dimensional scatter plots describing the accumulation of the sequences and their belonging to a specific cluster. The program does not have a graphical interface, and, if the user is using a non-Apple Macintosh operating system, the source code must be compiled before installation.
The significantly reduced pool, sized to tens or hundreds of sequences, can be subjected to the next stage of analysis based on the primary structure: the search for text motifs. For these purposes, the programs of the MEME Suite project [37] adapted for motif searching and analysis (MEME or GLAM) are the most often used. However, the programs do not predict the secondary structure of aptamers. The search for text motifs of various lengths among all sequences without prior data reduction processing can also be performed with the program MPBind [24], which evaluates the relative change in frequency and accumulation to the last rounds for all n-mer motifs. However, the program is written for Unix operating systems and requires advanced computer skills.
Folding structural approaches provide more complete information regarding the possible interaction of oligonucleotides with the target. These were applied earlier, when single sequences were randomly selected from the entire pool that successfully bound to the target [38]. However, in the context of HTS-SELEX analysis, a large-scale search for folding motifs is particularly relevant. In the course of library enrichment, the same folding structure can successfully accumulate; however, if it consists of fragments with different sizes or locations in a primary structure, text aligners would ignore this structure. However, most of the published folding searching approaches possess significant limitations.
Typically, they identify supposedly single-stranded regions (for instance AptaTRACE, which is a part of AptaSUITE described below) equating to a target binding site, and do not recognize other variants that may also be critical for complex formations. A successful motif search depends on the fine tuning of the parameters as biologically active folding is not always the most energetically favorable structure and can be stabilized by its target. In addition, such programs are focused on RNA libraries that can also distort the analysis if the library consists of oligonucleotides with other structures. The constant sequence regions should not be deleted or masked for the correct usage of a folding searching program as they can contribute to the formation of the secondary structure [39].
One of the first programs to search for aptamer folding motifs was Aptamotif [40]. The analysis is carried out in three stages: first, the optimal and suboptimal secondary structures are calculated using the RNAsubopt algorithm [41], then, the discovered structural fragments are extracted, successfully aligned fragments are selected for the next stage, and the most efficient variants are used to analyze the entire sequence library. The user needs to select the energy range of possible conformations to maintain a balance between the sensitivity and computational complexity. Another limitation is that the pool must be sufficiently enriched in binding aptamers—from 50% or more—since the algorithm does not equalize the entire pool, but only some of its random samples. Therefore, this approach may not give positive results for selection experiments with a high background binding level or should be used for datasets where non-specific sequences have been excluded.
APTANI was developed on the basis of Aptamotif [23]. In addition to analyzing secondary structures, it realizes some other functions such as aptamer clusterization by comparing the results of different HTS-SELEX cycles and searching for specific binding motifs. The updated version of the program, APTANI2 [42], has a graphical interface and provides more complete information regarding the enriched aptamers; moreover, it is capable of identifying a new variety of secondary structures including a rather important folding into G-quadruplex.
Other programs conduct comprehensive analysis of HTS-SELEX data using several different approaches to identify aptamers. For example, AptCompare [43] is a combination of AptaCluster [35], FASTAptamer [32], MPBind [24], APTANI [23], RNAmotifAnalysis [44], and a number of other scripts for preprocessing.
The proprietary software COMPAS performs complex data processing [45] and conducts analysis and clustering with different approaches that can be applied: comparison of full-length sequences or their 4–8 nucleotide k-mers, and comparison by the criteria of Shannon information entropy. The families grouped on the basis of the primary structure are combined with each other to form superfamilies according to possible similarities in the secondary structure. The program checks the input data and partially preprocesses them.
RaptRanker [46] also carries out complex data analysis. In the first stage, it passes all sequences through a rough filter, however, they must exactly match in length and in the sequence of constant regions. For unique sequences that have passed the preprocessing filter, a sequence structural profile is determined using the CapR algorithms [47], and clustering is performed according to a sub-sequence structural profile. The selection of specific candidates is based on the average motif enrichment that depends on the frequency of each subsequence, motif frequency, and motif enrichment for each cluster.
Finally, AptaSUITE [48] is a multifunctional program and a combination of previously published programs that carry out various stages of the analysis of HTS-SELEX data such as the previously mentioned AptaPLEX, AptaCLUSTER, and AptaTRACE as well as a program identifying polymerase errors AptaMUT [22] and simulator of HTS-SELEX—AptaSIM [48]. The AptaSIM program can be used for both adjusting the SELEX parameters and for an estimation of the influence of various factors on a real selection.
The program AptaPLEX [30] is used for data preprocessing. It sorts the obtained sequences according to barcodes into separate cycles, identifies and removes primers, and automatically corrects mismatches between forward and reverse reading. The combination of the programs analyzing the primary structure AptaCLUSTER and AptaMUT describes the dynamics of the pool enrichment, the representation of various clusters during the selection, and the accumulation of mutations in the clusters and their possible effect on the ability to bind to the target. The folding analysis is carried out by the AptaTRACE program, which indicates structures such as hairpins, bulge loops, inner loops, multiple loops, and dangling ends and evaluates their enrichment during selection. AptaSUITE is a cross-platform program and has both GUI and command line interfaces that make it convenient for people with different IT competencies.
In summary, the following different strategies can be outlined for the recognition of candidate aptamer sequences. The first is the abundance criteria. This approach arises from the initial basis of the SELEX experiment, which presumes the accumulation and enrichment of target specific sequences. The estimation of the enrichment rate from one selection cycle to another is a second approach for the discovery of aptamer sequences. This strategy not only accounts for the abundance, but also for the persistence of the individual sequence throughout the evolution process, and can help distinguish between the target-induced enrichment and biased sequence accumulation due to other non-specific reasons.
The third strategy relies on clusterization of the sequence dataset. In this case, prominent candidates are selected from different sequence clusters. As a result, less abundant but effective binders can be identified, while the sequences with the highest abundance and enrichment rate scores can originate from similar sequences with a moderate binding ability, which appear to be overrepresented due to stochastic reasons or selection bias. Finally, one further strategy for aptamer identification originates from the notion that not the entire oligonucleotide sequence but rather a rather short region exerts target binding; based on this, the enrichment of binding motifs instead of long sequences should serve for the recognition of aptamer sequences.
This strategy requires, first, the discovery of overrepresented binding motifs, which may both be structural sequences. The extracted motifs can be further clustered to achieve more precise aptamer identification. No systematic comparison of different strategies and software solutions for candidate aptamer identification is available at the moment; however, in certain cases, research articles devoted to the development of particular bioinformatics tools describe a comparison to other similar instruments [23][24][46].
Any of the described strategies or a combination of them can be applied in practical SELEX experiments. So far, the choice of software tools for the analysis of HTS experimental results depends primarily on the desired aptamer identification strategy. The computer competence of the researcher and the available user operating system can influence the software choice. Abundance scoring can be executed using almost all described software packages.
The cycle-to-cycle enrichment rate can be assessed using Galaxy, FASTAptamer, AptaSUITE (AptaPLEX), and RaptRankler. Cluster analysis can performed using ASTAptamer, AptaSUITE (AptaCluster), and SEWAL. A clusterization function is also available with Galaxy, but it is not adapted for SELEX experiments. Sequence motifs can be extracted using FASTAptamer, AptaSUITE, MPBind, MEME/GLAM (available with Galaxy or separately), and Tallymer. Structural motifs can be identified using APTANI, APTANI2, AptaSUITE (AptaTRACE), and RaptRanker. APTANI, MPBind, and RaptRanker are intended to provide rankings of aptamer candidates with prospective binding ability, thus, practically allows the user to escape rendering a final decision on candidate selection.
In relation to computer skills requirements, Galaxy is an absolutely outstanding tool due to its simplicity and availability for all users. The FASTAptamer and AptaSUITE packages require minimal user experience for the installation and command line operation. The execution of APTANI, APTANI2, RaptRanker, SEWAL, or MPBind requires more complex installation. APTANI and APTANI2 also demand some bioinformatics experience of the researcher due to the need to apply user-defined parameters. The functionality and characteristic features of the different HTS-SELEX software tools are summarized in Table 1.
Table 1. Features of the software tools developed or adapted for high-throughput sequencing data analysis in systematic evolution of ligands by exponential enrichment.
|
Software |
Analysis Stages |
Platforms |
GUI |
Source |
|
Galaxy |
Data preprocessing |
Web/Linux/Mac OS |
Yes |
https://usegalaxy.org/ |
|
Enrichment counting |
||||
|
FastAptamer |
Enrichment counting |
Linux/Mac OS/Windows/Galaxy |
No |
https://github.com/FASTAptamer/FASTAptamer |
|
Sequence based clustering |
||||
|
Searching known motifs |
||||
|
PATTERNITY-seq |
Sequence based clustering |
Linux/Mac OS/Windows |
No |
https://github.com/AptaFred/EGE_tree |
|
MEME/GLAM |
Sequence motif searching |
Linux/Mac OS/Web |
Yes (Web) |
http://meme-suite.org |
|
MPBind |
Data preprocessing |
Linux/Mac OS |
No |
https://morgridge.org/research/regenerative-biology/software-resources/mpbind/ |
|
Sequence motif searching |
||||
|
Aptamotif |
Structure motif searching |
Linux/Mac OS |
No |
By request |
|
APTANI |
Enrichment counting |
Linux/Mac OS |
No |
http://aptani.unimore.it/ |
|
Structure motif searching |
||||
|
APTANI2 |
Enrichment counting |
Linux/Mac OS |
Yes |
http://aptani.unimore.it/ |
|
Structure motif searching |
||||
|
AptCompare |
Data preprocessing |
Linux/Mac OS/Windows |
Yes |
https://bitbucket.org/shiehk/aptcompare/src/master/ |
|
Enrichment counting |
||||
|
Sequence based clustering |
||||
|
Sequence motif searching |
||||
|
Structure motif searching |
||||
|
COMPAS |
Data preprocessing |
Unknown |
Yes |
Unknown |
|
Enrichment counting |
||||
|
Sequence based clustering |
||||
|
Structure motif searching |
||||
|
RaptRanker |
Data preprocessing |
Linux/Mac OS |
No |
https://github.com/hmdlab/RaptRanker |
|
Enrichment counting |
||||
|
Structure motif searching |
||||
|
AptaSUITE |
Data preprocessing |
Linux/Mac OS/Windows |
Yes |
https://github.com/drivenbyentropy/aptasuite |
|
Enrichment counting |
||||
|
Sequence based clustering |
||||
|
Structure motif searching |
||||
|
SELEX simulation |
References
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510, doi:10.1126/science.2200121.
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822, doi:10.1038/346818a0.
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598, doi:10.3390/molecules24193598.
- Zhu, C.; Yang, G.; Ghulam, M.; Li, L.; Qu, F. Evolution of multi-functional capillary electrophoresis for high-efficiency selection of aptamers. Biotechnol. Adv. 2019, 37, 107432, doi:10.1016/j.biotechadv.2019.107432.
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351.
- Sharma, T.K.; Bruno, J.G.; Dhiman, A. ABCs of DNA aptamer and related assay development. Biotechnol. Adv. 2017, 35, 275–301, doi:10.1016/j.biotechadv.2017.01.003.
- Zimmermann, B.; Bilusic, I.; Lorenz, C.; Schroeder, R. Genomic SELEX: A discovery tool for genomic aptamers. Methods 2010, 52, 125–132, doi:10.1016/j.ymeth.2010.06.004.
- Van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20, doi:10.1016/j.yexcr.2014.01.008.
- Pawluczyk, M.; Weiss, J.; Links, M.G.; Egaña Aranguren, M.; Wilkinson, M.D.; Egea-Cortines, M. Quantitative evaluation of bias in PCR amplification and next-generation sequencing derived from metabarcoding samples. Anal. Bioanal. Chem. 2015, 407, 1841–1848, doi:10.1007/s00216-014-8435-y.
- Tolle, F.; Mayer, G. Preparation of SELEX Samples for Next-Generation Sequencing. In Nucleic Acid Aptamers. Methods in Molecular Biology, vol 1380; Mayer, G., Ed.; Humana Press, New York, NY, USA, 2016; pp. 77–84.
- Riley, K.R.; Gagliano, J.; Xiao, J.; Libby, K.; Saito, S.; Yu, G.; Cubicciotti, R.; Macosko, J.; Colyer, C.L.; Guthold, M.; et al. Combining capillary electrophoresis and next-generation sequencing for aptamer selection. Anal. Bioanal. Chem. 2015, 407, 1527–1532, doi:10.1007/s00216-014-8427-y.
- Valenzano, S.; De Girolamo, A.; DeRosa, M.C.; McKeague, M.; Schena, R.; Catucci, L.; Pascale, M. Screening and Identification of DNA Aptamers to Tyramine Using in Vitro Selection and High-Throughput Sequencing. ACS Comb. Sci. 2016, 18, 302–313, doi:10.1021/acscombsci.5b00163.
- Quang, N.N.; Miodek, A.; Cibiel, A.; Ducongé, F. Selection of Aptamers Against Whole Living Cells: From Cell-SELEX to Identification of Biomarkers. In Synthetic Antibodies; Humana Press: New York, NY, USA, 2017; pp. 253–272.
- Martin, J.A.; Parekh, P.; Kim, Y.; Morey, T.E.; Sefah, K.; Gravenstein, N.; Dennis, D.M.; Tan, W. Selection of an Aptamer Antidote to the Anticoagulant Drug Bivalirudin. PLoS ONE 2013, 8, e57341, doi:10.1371/journal.pone.0057341.
- Xu, J.; Teng, I.-T.; Zhang, L.; Delgado, S.; Champanhac, C.; Cansiz, S.; Wu, C.; Shan, H.; Tan, W. Molecular Recognition of Human Liver Cancer Cells Using DNA Aptamers Generated via Cell-SELEX. PLoS ONE 2015, 10, e0125863, doi:10.1371/journal.pone.0125863.
- Kimoto, M.; Matsunaga, K.; Hirao, I. DNA Aptamer Generation by Genetic Alphabet Expansion SELEX (ExSELEX) Using an Unnatural Base Pair System. In Nucleic Acid Aptamers; Humana Press: New York, NY, USA, 2016; pp. 47–60.
- Spiga, F.M.; Maietta, P.; Guiducci, C. More DNA–Aptamers for Small Drugs: A Capture–SELEX Coupled with Surface Plasmon Resonance and High-Throughput Sequencing. ACS Comb. Sci. 2015, 17, 326–333, doi:10.1021/acscombsci.5b00023.
- Hoon, S.; Zhou, B.; Janda, K.; Brenner, S.; Scolnick, J. Aptamer selection by high-throughput sequencing and informatic analysis. Biotechniques 2011, 51, 413–416, doi:10.2144/000113786.
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378, doi:10.1073/pnas.1009331107.
- Kupakuwana, G.V.; Crill, J.E.; McPike, M.P.; Borer, P.N. Acyclic Identification of Aptamers for Human alpha-Thrombin Using Over-Represented Libraries and Deep Sequencing. PLoS ONE 2011, 6, e19395, doi:10.1371/journal.pone.0019395.
- Beier, R.; Boschke, E.; Labudde, D. New Strategies for Evaluation and Analysis of SELEX Experiments. Biomed Res. Int. 2014, 2014, 1–12, doi:10.1155/2014/849743.
- Hoinka, J.; Berezhnoy, A.; Dao, P.; Sauna, Z.E.; Gilboa, E.; Przytycka, T.M. Large scale analysis of the mutational landscape in HT-SELEX improves aptamer discovery. Nucleic Acids Res. 2015, 43, 5699–5707, doi:10.1093/nar/gkv308.
- Caroli, J.; Taccioli, C.; De La Fuente, A.; Serafini, P.; Bicciato, S. APTANI: A computational tool to select aptamers through sequence-structure motif analysis of HT-SELEX data. Bioinformatics 2015, 32, 161–164, doi:10.1093/bioinformatics/btv545.
- Jiang, P.; Meyer, S.; Hou, Z.; Propson, N.E.; Soh, H.T.; Thomson, J.A.; Stewart, R. MPBind: A Meta-motif-based statistical framework and pipeline to Predict Binding potential of SELEX-derived aptamers. Bioinformatics 2014, 30, 2665–2667, doi:10.1093/bioinformatics/btu348.
- Jing, M.; Bowser, M.T. Tracking the Emergence of High Affinity Aptamers for rhVEGF 165 During Capillary Electrophoresis-Systematic Evolution of Ligands by Exponential Enrichment Using High Throughput Sequencing. Anal. Chem. 2013, 85, 10761–10770, doi:10.1021/ac401875h.
- Kurtz, S.; Narechania, A.; Stein, J.C.; Ware, D. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics 2008, 9, 517, doi:10.1186/1471-2164-9-517.
- Stoltenburg, R.; Nikolaus, N.; Strehlitz, B. Capture-SELEX: Selection of DNA Aptamers for Aminoglycoside Antibiotics. J. Anal. Methods Chem. 2012, 2012, 415697, doi:10.1155/2012/415697.
- Thiel, W.H.; Giangrande, P.H. Analyzing HT-SELEX data with the Galaxy Project tools—A web based bioinformatics platform for biomedical research. Methods 2016, 97, 3–10, doi:10.1016/j.ymeth.2015.10.008.
- Thiel, W.H. Galaxy Workflows for Web-based Bioinformatics Analysis of Aptamer High-throughput Sequencing Data. Mol. Ther. Nucleic Acids 2016, 5, e345, doi:10.1038/mtna.2016.54.
- Hoinka, J.; Przytycka, T. AptaPLEX—A dedicated, multithreaded demultiplexer for HT-SELEX data. Methods 2016, 106, 82–85, doi:10.1016/j.ymeth.2016.04.011.
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX Process with Next-Generation Sequencing. PLoS ONE 2011, 6, e29604, doi:10.1371/journal.pone.0029604.
- Alam, K.K.; Chang, J.L.; Burke, D.H. FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol. Ther. Nucleic Acids 2015, 4, e230, doi:10.1038/mtna.2015.4.
- Toulmé, J.-J.; Azéma, L.; Darfeuille, F.; Dausse, E.; Durand, G.; Paurelle, O. Aptamers in Bordeaux 2017: An exceptional “millésime.” Biochimie 2018, 145, 2–7, doi:10.1016/j.biochi.2017.11.015.
- Buhler, J. Efficient large-scale sequence comparison by locality-sensitive hashing. Bioinformatics 2001, 17, 419–428, doi:10.1093/bioinformatics/17.5.419.
- Hoinka, J.; Berezhnoy, A.; Sauna, Z.E.; Gilboa, E.; Przytycka, T.M. AptaCluster—A Method to Cluster HT-SELEX Aptamer Pools and Lessons from Its Application. In International Conference on Research in Computational Molecular Biology; Springer: Cham, Switzerland, 2014; pp. 115–128.
- Pitt, J.N.; Rajapakse, I.; Ferre-D’Amare, A.R. SEWAL: An open-source platform for next-generation sequence analysis and visualization. Nucleic Acids Res. 2010, 38, 7908–7915, doi:10.1093/nar/gkq661.
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208, doi:10.1093/nar/gkp335.
- Lozupone, C.; Changayil, S.; Majerfeld, I.; Yarus, M. Selection of the simplest RNA that binds isoleucine. RNA 2003, 9, 1315–1322, doi:10.1261/rna.5114503.
- Cowperthwaite, M.C.; Ellington, A.D. Bioinformatic Analysis of the Contribution of Primer Sequences to Aptamer Structures. J. Mol. Evol. 2008, 67, 95–102, doi:10.1007/s00239-008-9130-4.
- Hoinka, J.; Zotenko, E.; Friedman, A.; Sauna, Z.E.; Przytycka, T.M. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 2012, 28, i215–i223, doi:10.1093/bioinformatics/bts210.
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26, doi:10.1186/1748-7188-6-26.
- Caroli, J.; Forcato, M.; Bicciato, S. APTANI2: Update of aptamer selection through sequence-structure analysis. Bioinformatics 2020, 36, 2266–2268, doi:10.1093/bioinformatics/btz897.
- Shieh, K.R.; Kratschmer, C.; Maier, K.E.; Greally, J.M.; Levy, M.; Golden, A. AptCompare: Optimized de novo motif discovery of RNA aptamers via HTS-SELEX. Bioinformatics 2020, 36, 2905–2906, doi:10.1093/bioinformatics/btaa054.
- Ditzler, M.A.; Lange, M.J.; Bose, D.; Bottoms, C.A.; Virkler, K.F.; Sawyer, A.W.; Whatley, A.S.; Spollen, W.; Givan, S.A.; Burke, D.H. High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of HIV reverse transcriptase. Nucleic Acids Res. 2013, 41, 1873–1884, doi:10.1093/nar/gks1190.
- Blank, M. Next-Generation Analysis of Deep Sequencing Data: Bringing Light into the Black Box of SELEX Experiments. In Nucleic Acid Aptamers. Selection, Characterization, and Application; Humana Press: New York, NY, USA, 2016; pp. 85–95.
- Ishida, R.; Adachi, T.; Yokota, A.; Yoshihara, H.; Aoki, K.; Nakamura, Y.; Hamada, M. RaptRanker: In silico RNA aptamer selection from HT-SELEX experiment based on local sequence and structure information. Nucleic Acids Res. 2020, 48, e82, doi:10.1093/nar/gkaa484.
- Fukunaga, T.; Ozaki, H.; Terai, G.; Asai, K.; Iwasaki, W.; Kiryu, H. CapR: Revealing structural specificities of RNA-binding protein target recognition using CLIP-seq data. Genome Biol. 2014, 15, R16, doi:10.1186/gb-2014-15-1-r16.
- Hoinka, J.; Backofen, R.; Przytycka, T.M. AptaSUITE: A Full-Featured Bioinformatics Framework for the Comprehensive Analysis of Aptamers from HT-SELEX Experiments. Mol. Ther. Nucleic Acids 2018, 11, 515–517, doi:10.1016/j.omtn.2018.04.006.

