 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xinxin Xiao | + 2017 word(s) | 2017 | 2021-01-27 11:47:11 | | | |
| 2 | Peter Tang | -2 word(s) | 2015 | 2021-02-02 04:24:45 | | | | |
| 3 | Xinxin Xiao | Meta information modification | 2015 | 2021-02-08 11:43:09 | | |
Video Upload Options
Self-assembled molecular monolayers (SAMs) are long recognized as crucial "bridges" between redox enzymes and solid electrode surfaces, on which the enzymes undergo direct electron transfer (DET)—for example, in enzymatic biofuel cells (EBFCs) and biosensors. SAMs possess a wide range of terminal groups that enable productive enzyme adsorption and fine-tuning in favorable orientations on the electrode. The tunneling distance, the SAM chain length, and the contacting terminal SAM groups are the most significant factors in SAM-controlled DET-type bioelectrocatalysis.
1. Introduction
Self-assembled molecular monolayers (SAMs) are surface monolayers that spontaneously bind to metal surfaces on which, for example, the unique metal-S bonding between metal and thiols offers a versatile pathway to tailor interfacial electrochemical and bioelectrochemical properties [1][2][3][4]. Thiol SAMs on metal surfaces are core targets to provide an understanding of self-organization and interfacial interactions at the molecular level in biological systems [5][6]. Bigelow and associates were the first to demonstrate well-oriented SAMs adsorbed on a platinum wire [7], but SAMs did not attract much attention until Nuzzo and associates discovered disulfide monolayers on gold substrates in solution, as a phenomenon different from conventional Langmuir–Blodgett (LB) films [8]. Besides gold and platinum, SAMs can also form on the surfaces of other metals including silver, copper, palladium, and mercury [9][10][11][12][13][14]. Gold is, however, the most extensively investigated because of its chemical inertness, relatively easy handling, and wide potential window, suitable for a range of electrochemical studies [1][15].
In parallel, the nature of the surface Au-S bond has been in intense focus over a number of years [1][2][4] and was recently overviewed [16][17]. In contrast to molecular Au(I)-S(-I) complexes, the "aurophilic" effect arising from collective interactions among surface Au atoms leads to displacement of the 6s Au electrons out of chemical reach and the filled 5d electrons taking over in Au-S bonding. The Au-S bond on Au surfaces is thus an intriguing example of strong, aurophilically controlled van der Waals binding in an Au(0)-•S(0) gold(0)-thiyl bond.
Electron transfer (ET) reactions between an electrode surface and an oxidoreductase is the core "elementary" process in bioelectrocatalysis [18][19][20][21][22]. For example, oxidoreductases possess redox/catalytic center(s) that catalyze the oxidation of fuels on a bioanode (e.g., glucose, fructose, lactate, and sulfite) [20][23][24][25], which can be assembled with a biocathode undergoing biocatalytic reduction reactions, typically dioxygen reduction, in enzymatic biofuel cells (EBFCs) [26][27][28][29]. The bioelectrochemical ET processes are classified into direct ET (DET) and mediated ET (MET) [19][20][30]. MET utilizes external and artificial redox mediators to shuttle the electrons between the electrode and the oxidoreductase, especially if the redox center(s) are buried deep inside the protein structure [31][32]. In DET, redox enzymes communicate directly with the electrode surface, if the redox cofactors/centers are spatially close to the electrode surfaces (generally less than 2 nm), facilitating electron tunneling [33]. DET is thus a simpler mechanism by eliminating the need for external redox mediators, and it is therefore amenable to more detailed mechanistic analysis.
Oxidoreductase immobilization is crucial to improving electrode reusability and stability [19]. To achieve efficient DET, the detailed surface characteristics of both enzyme and electrode for favorable enzyme orientation must be considered, leading to shortest possible electron tunneling distance. A wide range of carbon or metallic supports have been employed for effective enzyme immobilization [25][28][34][35][36][37]. Physical adsorption and covalent bonding are most commonly used. SAMs have been introduced to serve as a bridge for gentle protein/enzyme immobilization on gold or other metal surfaces [36][38][39]. SAM structures are determined by the Au-S bond, the local surface structure of the metal surfaces, lateral interactions, as well as by the solvent and electrolytes [6]. Use of SAMs avoids the direct contact of enzyme and solid surfaces [40], and mimicks the microenvironment in biological membranes. SAMs hold a variety of functional hydrophilic or hydrophobic terminal groups, such as carboxyl, hydroxyl, amino, and alkyl groups [41]. Frequently used alkanethiol and thiophenols are summarized in Figure 1. In addition, the DET kinetics is controlled by the SAM molecular chain length, as the ET rate depends strongly on the tunneling distance [42][43][44]. Emerging protein engineering provides oxidoreductases directly with thiol residues, leading to controlled orientation either by direct protein thiol binding or by thioether bond formation with unsaturated maleimide on the surface [45].
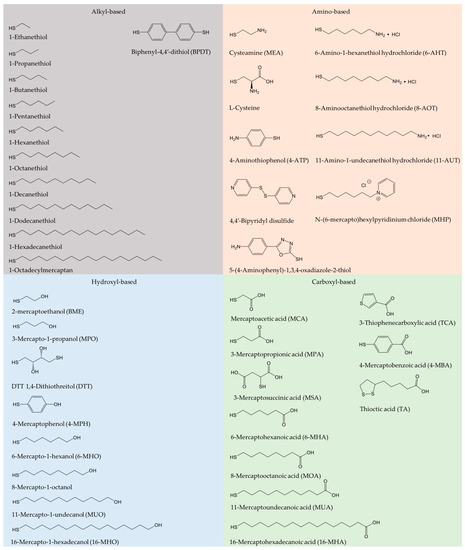
Figure 1. Frequently used alkanethiols and thiophenols in the formation of self-assembled molecular monolayers (SAMs) with alkyl, amino, hydroxyl, and carboxyl terminal groups.
Current density and enzyme loading can be promoted using nanostructured materials [3][44][46][47][48]. Among these, metallic nanomaterials exhibit excellent electronic conductivity and large surface area, with clear potential for improved catalytic response and stability of immobilized redox enzymes [25]. Nanoporous gold (NPG), prepared via de-alloying Au alloys or electrodeposition, with three-dimensional porous architecture and relatively uniform pore size, is a particular candidate for immobilizing enzymes in DET [25][28][32][49][50]. Gold nanoparticles (AuNPs) featuring a spherical nanostructure and large surface areas have also been widely studied in bioelectrocatalysis [51][52][53][54]. The combination of SAMs and nanostructured gold offers new opportunities in bioelectrochemistry and has been extensively reviewed [21][32][55], but only a few reviews cover SAMs on planar or porous gold electrodes for controlled enzyme orientation [3][19]. DET on carbon electrode materials (carbon nanotubes, graphene-based materials) is another major parallel sector, beyond the scope of the present focused review but recently reviewed elsewhere [18][56][57][58].
2. Preparation and Characterization of SAM-Based Au- and Other Electrodes
2.1. Preparation of SAMs
Thiol SAMs form spontaneously on electronically soft metal substrates such as gold surfaces from liquid or vapor media under mild conditions [5][59]. Well-defined SAMs are formed by immersing the clean metal surface into a thiol solution, commonly ethanol or aqueous solution, for a certain period of time (hours to days), followed by washing with the same solvent. The SAM quality is determined by the variety and concentration of thiol solution, temperature, soaking time, and the metal surface structure. Vapor deposition is also used to prepare SAMs, but the SAM morphology is here hard to control. Single-crystal, atomically planar gold electrodes are the best substrates for SAM studies, companied by characterization by several in situ techniques including electrochemical scanning tunneling microscopy (in situ STM). Nanomaterials with porous structure and high surface area are also used as substrates [40][60], and a potential-assisted method to accelerate the SAM formation process has been developed [9][61][62]. The electrochemical potential range is then a key parameter that strongly affects the quality of the SAMs, evaluated for example by the surface coverage.
2.2. Characterization Methods
2.2.1. Electrochemistry
SAMs can be characterized by a range of methods, among which electrochemical methods have been widely reported [59]. Electrochemical methods involve cyclic (CV) and linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS). CV of non-redox active SAMs, i.e., capacitive voltammetry, generally shows decreased double-layer capacitance after SAM adsorption on a metal surface. LSV and CV records the irreversible reductive (and oxidative) desorption of SAMs at a negative (or positive) potential. Reductive desorption is well suited to evaluate the surface coverage and the thermodynamic stability of the SAM Au-S units. The shape and position of the desorption peaks are sensitive to the crystalline surface structure and the SAM species. EIS can disentangle the ET resistance from the mass transfer or diffusion resistance. The ET resistance increases with increasing chain length because of the increased tunneling distance [63]. Electrochemical quartz crystal microbalance (EQCM) is an in situ technique that allows real-time monitoring of the SAM adsorption process with high sensitivity to the mass changes on the electrode [59].
2.2.2. Microscopy
Electrochemical scanning probe microscopies (SPMs), especially STM and atomic force microscopy (AFM), have been extensively used to provide structural SAM properties at the single-molecule level. STM rests on the quantum tunneling effect. A bias voltage is applied across the metal support and the extremely sharp STM probe, generating tunneling currents, transformed to high-resolution conductivity images as the STM tip is scanned across the SAM-modified metal support. Zhang and associates have reported extensive in situ STM investigations using a wide range of alkanethiols on single-crystal gold [6][64][65][66], recording high-resolution mapping of SAM thickness and molecular orientations. In situ STM combined with electrochemical control can also directly map real-time SAM evolution of structural features. In situ AFM has also attracted attention in bioelectrochemistry [67]. AFM records complex forces, mapping the surface structure by allowing the tip to directly contact the redox protein/enzyme molecules. Contact angle (CA) measurement is a simple and straightforward method for monitoring the hydrophobic/hydrophilic properties of SAMs [1][5].
2.2.3. Spectroscopy
Spectroscopic techniques are needed to map the complex interactions between SAMs and the metal support [16][17]. X-ray photoelectron spectroscopy (XPS) records the element composition and the chemical state of the SAMs. High-resolution XPS spectra of S 2p usually show a doublet at the binding energy ranging from 160 to 165 eV, attributed to the formation of the Au-S bond between alkanethiol molecules and the metal support. The functional SAM groups can be identified by Fourier-transform infrared spectroscopy (FTIR), and Raman spectroscopy can be used to detect structural changes in SAMs [68]. Surface plasmon resonance (SPR) spectroscopy is a powerful technique to measure the SAM thickness and shows a change in the tilt angle when the SAM thickness varies. Adsorption kinetics can be monitored online by SPR coupled with ellipsometry [62].
3. SAMs and Electrochemical DET of Redox Proteins/Enzymes
Redox proteins and enzymes immobilized on solid surfaces are fragile biomolecular entities. With a few exceptions, they retain ET or enzyme function only in prepared microenvironments that somehow emulate their natural aqueous/membrane reaction media. With such perspectives SAMs have received considerable early and recent attention as substrates for interfacial bioelectrochemical redox protein and enzyme DET reactions. Self-assembled molecular monolayers with multifariously functionalized thiols are close to ideal SAM building blocks which have emerged as the core SAM class over the last couple of decades. The -SH thiol group ascertains robust Au-S linking, now increasingly well understood as dominated by the Au(0)-•S(0) gold-thiyl and strong van der Waals Au-S interaction. The opposite functionalized end of the SAM thiol molecules offers the choice of hydrophilic or hydrophobic, electrostatically charged or neutral, and structurally large or small terminal groups, well suited for designed gentle protein/enzyme linking to the solid SAM-modified electrochemical surface. The option of varying the length of the SAM-forming molecules offers additional control of the electron tunneling process, as well as of the dielectric and other local environmental properties crucial in the overall control of the electrochemical activity of the immobilized proteins or enzymes.
A wide range of metalloproteins have been investigated and their catalytic mechanism and ET kinetics on various SAM-modified electrode surfaces mapped in detail. The redox active centers of protein/enzymes in DET can be roughly categorized into two major groups: metal-based (iron, copper and molybdenum centers etc.) [20][69][70][71] and non-metal-based (e.g., flavin adenine dinucleotide (FAD) and pyrrolo-quinoline quinone (PQQ)) centers [44][72][73]. Most, although not all, DET-capable enzymes harbor multiple redox centers, with internal ET relays via heme groups, copper clusters, or Fe-S clusters, which shuttle electrons between the electrode surface and the catalytic cofactors [45][74][75]. Recent core studies where strategically chosen SAMs in DET-type electrocatalysis have been crucial, and mechanisms of interfacial DET mapped in detail are summarized in Figure 2 [4][15][19][32][64][67][70][76][77][78][79][80][81]. Particularly, these studies have focused on, how favorable enzyme orientations can be obtained using structurally versatile SAMs, and how the interactions between the redox protein/enzyme and the SAM-modified electrodes can be tuned. In these ways the studies show, how the impressive potential of well-chosen thiol-based SAMs ascertains robust performance and can lead to detailed molecular mechanistic mapping of the electrochemistry of large complex biomolecules.
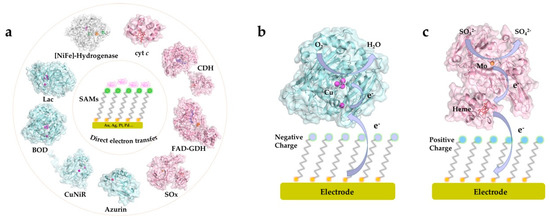
Figure 2. (a) Illustration of common proteins/enzymes capable of direct electron transfer (DET) on electrode modified by self-assembled molecular monolayers (SAMs). The proteins include: horse heart cytochrome c (cyt c), PDB 1HRC; cellobiose dehydrogenase (CDH) from Neurospora crassa, PDB 4QI7; gamma-alpha subunit of FAD-dependent glucose dehydrogenase (FAD-GDH) from Burkholderia cepacia, PDB 6A2U; chicken liver sulfite oxidase (SOx), PDB 1SOX; Pseudomonas aeruginosa azurin T30R1, PDB 5I28; monomer of Achromobacter xylosoxidans copper nitrite reductase (AxCuNiR), PDB 1HAU; bilirubin oxidase from Myrothecium verrucaria (BOD), PDB 2XLL; laccase (Lac) from Trametes versicolor, PDB 1KYA; [NiFe]-hydrogenase from DesulfovVibrio Vulgaris Miyazaki F (DvMF), PDB 1UBU. Schematic view of representative DET processes of (b) mvBOD and (c) SOx on the electrode modified with negatively and positively charged SAMs, respectively.
References
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 11031–170, doi.org/10.1021/cr0300789.
- Nöll, T.; Nöll, G. Strategies for “wiring” redox-active proteins to electrodes and applications in biosensors, biofuel cells, and nanotechnology. Chem. Soc. Rev. 2011, 40, 3564–3576, doi:10.1039/c1cs15030h.
- Bollella, P. Porous Gold: A New Frontier for Enzyme-Based Electrodes. Nanomaterials 2020, 10, 722, doi:10.3390/nano10040722.
- Chi, Q.; Ford, M.J.; Halder, A.; Hush, N.S.; Reimers, J.R.; Ulstrup, J. Sulfur ligand mediated electrochemistry of gold surfaces and nanoparticles: What, how, and why. Curr. Opin. Electrochem. 2017, 1, 7–15, doi:10.1016/j.coelec.2016.12.004.
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev. 1996, 96, 1533–1554, doi:10.1021/cr9502357.
- Zhang, J.; Kuznetsov, A.M.; Medvedev, I.G.; Chi, Q.; Albrecht, T.; Jensen, P.S.; Ulstrup, J. Single-molecule electron transfer in electrochemical environments. Chem. Rev. 2008, 108, 2737–2791, doi:10.1021/cr068073+.
- Bigelow, W.C.; Pickett, D.L.; Zisman, W.A. Oleophobic monolayers. I. Films adsorbed from solution in non-polar liquids. J. Colloid Sci. 1946, 1, 513–538, doi:10.1016/0095-8522(46)90059-1.
- Nuzzo, R.G.; Allara, D.L. Adsorption of Bifunctional Organic Disulfides on Gold Surfaces. J. Am. Chem. Soc. 1983, 105, 4481–4483, doi:10.1021/ja00351a063.
- Muskal, N.; Mandler, D. Thiol self-assembled monolayers on mercury surfaces : The adsorption and electrochemistry of v-mercaptoalkanoic acids. Electrochim. Acta 1999, 45, 537–548, doi.org/10.1016/S0013-4686(99)00232-7.
- Williams, J.A.; Gorman, C.B. Alkanethiol Reductive Desorption from Self-Assembled Monolayers on Gold, Platinum, and Palladium Substrates. J. Phys. Chem C 2007, 111, 12804–12810, doi:10.1021/jp072869a.
- Hoque, E.; Derose, J.A.; Houriet, R.; Hoffmann, P.; Mathieu, H.J.; Fe, E.P. Stable Perfluorosilane Self-Assembled Monolayers on Copper Oxide Surfaces: Evidence of Siloxy-Copper Bond Formation. Chem. Mater. 2007, 19, 798–804, doi:10.1021/cm062318h.
- Fischer, S.; Papageorgiou, A.C.; Marschall, M.; Reichert, J.; Diller, K.; Klappenberger, F.; Allegretti, F.; Nefedov, A.; Wöll, C.; Barth, J.V. L-Cysteine on Ag(111): A combined STM and X-ray spectroscopy study of anchorage and deprotonation. J. Phys. Chem. C 2012, 116, 20356–20362, doi:10.1021/jp305270h.
- Alejandra, M.; Addato, F.; Rubert, A.A.; Benítez, G.A.; Fonticelli, M.H.; Carrasco, J.; Carro, P.; Salvarezza, R.C. Alkanethiol Adsorption on Platinum: Chain Length Effects on the Quality of Self-Assembled Monolayers. J. Phys. Chem. C 2011, 115, 17788–17798, doi:10.1021/jp201390m.
- Feliciano-ramos, I.; Caban-acevedo, M.; Scibioh, M.A.; Cabrera, C.R. Self-assembled monolayers of L-cysteine on palladium electrodes. J. Electroanal. Chem. 2010, 650, 98–104, doi:10.1016/j.jelechem.2010.09.001.
- Gooding, J.J.; Ciampi, S. The molecular level modification of surfaces: From self-assembled monolayers to complex molecular assemblies. Chem. Soc. Rev. 2011, 40, 2704–2718, doi:10.1039/c0cs00139b.
- Reimers, J.R.; Ford, M.J.; Marcuccio, S.M.; Ulstrup, J.; Hush, N.S. Competition of van der Waals and chemical forces on gold-sulfur surfaces and nanoparticles. Nat. Rev. Chem. 2017, 1, 1–13, doi:10.1038/s41570-017-0017.
- Reimers, J.R.; Ford, M.J.; Halder, A.; Ulstrup, J.; Hush, N.S. Gold surfaces and nanoparticles are protected by Au(0)-thiyl species and are destroyed when Au(I)-thiolates form. Proc. Natl. Acad. Sci. USA 2016, 113, E1424–E1433, doi:10.1073/pnas.1600472113.
- Tang, J.; Yan, X.; Engelbrekt, C.; Ulstrup, J.; Magner, E.; Xiao, X.; Zhang, J. Development of graphene-based enzymatic biofuel cells: A minireview. Bioelectrochemistry 2020, 134, 107537, doi:10.1016/j.bioelechem.2020.107537.
- Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; de Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling redox enzyme orientation at planar electrodes. Catalysts 2018, 8, 192, doi:10.3390/catal8050192.
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253, doi:10.1098/rsif.2017.0253.
- Mano, N.; De Poulpiquet, A. O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468, doi:10.1021/acs.chemrev.7b00220.
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802, doi:10.1038/355796a0.
- Schulz, C.; Kittl, R.; Ludwig, R.; Gorton, L. Direct Electron Transfer from the FAD Cofactor of Cellobiose Dehydrogenase to Electrodes. ACS Catal. 2016, 6, 555–563, doi:10.1021/acscatal.5b01854.
- Lee, H.; Lee, Y.S.; Reginald, S.S.; Baek, S.; Lee, E.M.; Choi, I.G.; Chang, I.S. Biosensing and electrochemical properties of flavin adenine dinucleotide (FAD)-Dependent glucose dehydrogenase (GDH) fused to a gold binding peptide. Biosens. Bioelectron. 2020, 165, 112427, doi:10.1016/j.bios.2020.112427.
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The Immobilization of Fructose Dehydrogenase on Nanoporous Gold Electrodes for the Detection of Fructose. ChemElectroChem 2017, 4, 905–912, doi:10.1002/celc.201600842.
- Gutierrez-Sanchez, C.; Ciaccafava, A.; Blanchard, P.Y.; Monsalve, K.; Giudici-Orticoni, M.T.; Lecomte, S.; Lojou, E. Efficiency of Enzymatic O2 Reduction by Myrothecium verrucaria Bilirubin Oxidase Probed by Surface Plasmon Resonance, PMIRRAS, and Electrochemistry. ACS Catal. 2016, 6, 5482–5492, doi:10.1021/acscatal.6b01423.
- Di Bari, C.; Shleev, S.; De Lacey, A.L.; Pita, M. Laccase-modified gold nanorods for electrocatalytic reduction of oxygen. Bioelectrochemistry 2016, 107, 30–36, doi:10.1016/j.bioelechem.2015.10.003.
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Conghaile, P.; Pita, M.; Ludwig, R.; Magner, E. Immobilization of Redox Enzymes on Nanoporous Gold Electrodes: Applications in Biofuel Cells. ChemPlusChem 2017, 82, 553–560, doi:10.1002/cplu.201600455.
- Murata, K.; Kajiya, K.; Nakamura, N.; Ohno, H. Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell. Energy Environ. Sci. 2009, 2, 1280–1285, doi:10.1039/b912915d.
- Adachi, T.; Kaida, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Bioelectrocatalytic performance of D-fructose dehydrogenase. Bioelectrochemistry 2019, 129, 1–9, doi:10.1016/j.bioelechem.2019.04.024.
- Xiao, X.; Si, P.; Magner, E. An overview of dealloyed nanoporous gold in bioelectrochemistry. Bioelectrochemistry 2016, 109, 117–126, doi:10.1016/j.bioelechem.2015.12.008.
- Xiao, X.; Xia, H.Q.; Wu, R.; Bai, L.; Yan, L.; Magner, E.; Cosnier, S.; Lojou, E.; Zhu, Z.; Liu, A. Tackling the Challenges of Enzymatic (Bio)Fuel Cells. Chem. Rev. 2019, 119, 9509–9558, doi:10.1021/acs.chemrev.9b00115.
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236, doi:10.3390/catal10020236.
- Vaz-Dominguez, C.; Campuzano, S.; Rüdiger, O.; Pita, M.; Gorbacheva, M.; Shleev, S.; Fernandez, V.M.; De Lacey, A.L. Laccase electrode for direct electrocatalytic reduction of O2 to H2O with high-operational stability and resistance to chloride inhibition. Biosens. Bioelectron. 2008, 24, 531–537, doi:10.1016/j.bios.2008.05.002.
- Lindgren, A.; Gorton, L.; Ruzgas, T.; Baminger, U.; Haltrich, D.; Schülein, M. Direct electron transfer of cellobiose dehydrogenase from various biological origins at gold and graphite electrodes. J. Electroanal. Chem. 2001, 496, 76–81, doi:10.1016/S0022-0728(00)00251-5.
- Yan, Y.M.; Baravik, I.; Tel-Vered, R.; Willner, I. An ethanol/O2 biofuel cell based on an electropolymerized bilirubin oxidase/Pt nanoparticle bioelectrocatalytic O2-reduction cathode. Adv. Mater. 2009, 21, 4275–4279, doi:10.1002/adma.200900206.
- Schubert, K.; Goebel, G.; Lisdat, F. Bilirubin oxidase bound to multi-walled carbon nanotube-modified gold. Electrochim. Acta 2009, 54, 3033–3038, doi:10.1016/j.electacta.2008.12.010.
- Climent, V.; Zhang, J.; Friis, E.P.; Østergaard, L.H.; Ulstrup, J. Voltammetry and single-molecule in situ scanning tunneling microscopy of laccases and bilirubin oxidase in electrocatalytic dioxygen reduction on Au(111) single-crystal electrodes. J. Phys. Chem. C 2012, 116, 1232–1243, doi:10.1021/jp2086285.
- Chi, Q.; Farver, O.; Ulstrup, J. Long-range protein electron transfer observed at the single-molecule level: In situ mapping of redox-gated tunneling resonance. Proc. Natl. Acad. Sci. USA 2005, 102, 16203–16208, doi:10.1073/pnas.0508257102.
- Xiao, X.; Li, H.; Wang, M.; Zhang, K.; Si, P. Examining the effects of self-assembled monolayers on nanoporous gold based amperometric glucose biosensors. Analyst 2013, 139, 488–494, doi:10.1039/c3an01670f.
- Murata, K.; Suzuki, M.; Kajiya, K.; Nakamura, N.; Ohno, H. High performance bioanode based on direct electron transfer of fructose dehydrogenase at gold nanoparticle-modified electrodes. Electrochem. Commun. 2009, 11, 668–671, doi:10.1016/j.elecom.2009.01.011.
- Tominaga, M.; Ohtani, M.; Taniguchi, I. Gold single-crystal electrode surface modified with self-assembled monolayers for electron tunneling with bilirubin oxidase. Phys. Chem. Chem. Phys. 2008, 10, 6928–6934, doi:10.1039/b809737b.
- Chi, Q.; Zhang, J.; Andersen, J.E.T.; Ulstrup, J. Ordered assembly and controlled electron transfer of the blue copper protein azurin at gold (111) single-crystal substrates. J. Phys. Chem. B 2001, 105, 4669–4679, doi:10.1021/jp0105589.
- Kizling, M.; Dzwonek, M.; Więckowska, A.; Bilewicz, R. Size Does Matter—Mediation of Electron Transfer by Gold Clusters in Bioelectrocatalysis. ChemCatChem 2018, 10, 1988–1992, doi:10.1002/cctc.201800032.
- Al-Lolage, F.A.; Meneghello, M.; Ma, S.; Ludwig, R.; Bartlett, P.N. A Flexible Method for the Stable, Covalent Immobilization of Enzymes at Electrode Surfaces. ChemElectroChem 2017, 4, 1528–1534, doi:10.1002/celc.201700135.
- Matsumura, H.; Ortiz, R.; Ludwig, R.; Igarashi, K.; Samejima, M.; Gorton, L. Direct electrochemistry of phanerochaete chrysosporium cellobiose dehydrogenase covalently attached onto gold nanoparticle modified solid gold electrodes. Langmuir 2012, 28, 10925–10933, doi:10.1021/la3018858.
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225, doi:10.1016/j.bios.2011.10.020.
- Rauf, S.; Zhou, D.; Abell, C.; Klenerman, D.; Kang, D.J. Building three-dimensional nanostructures with active enzymes by surface templated layer-by-layer assembly. Chem. Commun. 2006, 1721–1723, doi:10.1039/b517557g.
- Xiao, X.; Siepenkoetter, T.; Conghaile, P.; Leech, D.; Magner, E. Nanoporous Gold-Based Biofuel Cells on Contact Lenses. ACS Appl. Mater. Interfaces 2018, 10, 7107–7116, doi:10.1021/acsami.7b18708.
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Belochapkine, S.; Magner, E. Nanoporous Gold Electrodes with Tuneable Pore Sizes for Bioelectrochemical Applications. Electroanalysis 2016, 28, 2415–2423, doi:10.1002/elan.201600249.
- Jensen, P.S.; Chi, Q.; Zhang, J.; Ulstrup, J. Long-Range interfacial electrochemical electron transfer of Pseudomonas aeruginosa azurin-gold nanoparticle hybrid systems. J. Phys. Chem. C 2009, 113, 13993–14000, doi:10.1021/jp902611x.
- Xu, S.; Han, X. A novel method to construct a third-generation biosensor: Self-assembling gold nanoparticles on thiol-functionalized poly(styrene-co-acrylic acid) nanospheres. Biosens. Bioelectron. 2004, 19, 1117–1120, doi:10.1016/j.bios.2003.09.007.
- Murata, K.; Suzuki, M.; Nakamura, N.; Ohno, H. Direct evidence of electron flow via the heme c group for the direct electron transfer reaction of fructose dehydrogenase using a silver nanoparticle-modified electrode. Electrochem. Commun. 2009, 11, 1623–1626, doi:10.1016/j.elecom.2009.06.012.
- Gutiérrez-Sánchez, C.; Pita, M.; Vaz-Domínguez, C.; Shleev, S.; De Lacey, A.L. Gold nanoparticles as electronic bridges for laccase-based biocathodes. J. Am. Chem. Soc. 2012, 134, 17212–17220, doi:10.1021/ja307308j.
- Meredith, M.T.; Minteer, S.D. Biofuel Cells: Enhanced Enzymatic Bioelectrocatalysis. Annu. Rev. Anal. Chem. 2012, 5, 157–179, doi:10.1146/annurev-anchem-062011-143049.
- Gross, A.J.; Holzinger, M.; Cosnier, S. Buckypaper bioelectrodes: Emerging materials for implantable and wearable biofuel cells. Energy Environ. Sci. 2018, 11, 1670–1687, doi:10.1039/c8ee00330k.
- Mazurenko, I.; de Poulpiquet, A.; Lojou, E. Recent developments in high surface area bioelectrodes for enzymatic fuel cells. Curr. Opin. Electrochem. 2017, 5, 74–84, doi:10.1016/j.coelec.2017.07.001.
- Mazurenko, I.; Hitaishi, V.P.; Lojou, E. Recent advances in surface chemistry of electrodes to promote direct enzymatic bioelectrocatalysis. Curr. Opin. Electrochem. 2020, 19, 113–121, doi:10.1016/j.coelec.2019.11.004.
- Chaki, N.K.; Vijayamohanan, K. Self-assembled monolayers as a tunable platform for biosensor applications. Biosens. Bioelectron. 2002, 17, 1–12, doi:10.1016/S0956-5663(01)00277-9.
- Xiao, X.; Ulstrup, J.; Li, H.; Wang, M.; Zhang, J.; Si, P. Nanoporous gold assembly of glucose oxidase for electrochemical biosensing. Electrochim. Acta 2014, 130, 559–567, doi:10.1016/j.electacta.2014.02.146.
- Serleti, A.; Salaj-Kosla, U.; Magner, E. The spatial and sequential immobilisation of cytochrome c at adjacent electrodes. Chem. Commun. 2013, 49, 8395–8397, doi:10.1039/c3cc44724c.
- Meunier-Prest, R.; Legay, G.; Raveau, S.; Chiffot, N.; Finot, E. Potential-assisted deposition of mixed alkanethiol self-assembled monolayers. Electrochim. Acta 2010, 55, 2712–2720, doi:10.1016/j.electacta.2009.12.045.
- Bradbury, C.R.; Zhao, J.; Fermín, D.J. Distance-independent charge-transfer resistance at gold electrodes modified by thiol monolayers and metal nanoparticles. J. Phys. Chem. C 2008, 112, 10153–10160, doi:10.1021/jp710165d.
- Chi, Q.; Zhang, J.; Arslan, T.; Borg, L.; Pedersen, G.W.; Christensen, H.E.M.; Nazmudtinov, R.R.; Ulstrup, J. Approach to interfacial and intramolecular electron transfer of the diheme protein cytochrome c4 assembled on Au(111) surfaces. J. Phys. Chem. B 2010, 114, 5617–5624, doi:10.1021/jp1007208.
- Engelbrekt, C.; Nazmutdinov, R.R.; Zinkicheva, T.T.; Glukhov, D.V.; Yan, J.; Mao, B.; Ulstrup, J.; Zhang, J. Chemistry of cysteine assembly on Au(100): Electrochemistry, in situ STM and molecular modeling. Nanoscale 2019, 11, 17235–17251, doi:10.1039/c9nr02477h.
- Zhang, J.; Welinder, A.C.; Chi, Q.; Ulstrup, J. Electrochemically controlled self-assembled monolayers characterized with molecular and sub-molecular resolution. Phys. Chem. Chem. Phys. 2011, 13, 5526–5545, doi:10.1039/c0cp02183k.
- Hao, X.; Zhang, J.; Christensen, H.E.M.; Wang, H.; Ulstrup, J. Electrochemical single-molecule AFM of the redox metalloenzyme copper nitrite reductase in action. ChemPhysChem 2012, 13, 2919–2924, doi:10.1002/cphc.201200220.
- Marmisollé, W.A.; Capdevila, D.A.; De La Llave, E.; Williams, F.J.; Murgida, D.H. Self-assembled monolayers of NH2-terminated thiolates: Order, pKa, and specific adsorption. Langmuir 2013, 29, 5351–5359, doi:10.1021/la304730q.
- Hitaishi, V.P.; Clément, R.; Quattrocchi, L.; Parent, P.; Duché, D.; Zuily, L.; Ilbert, M.; Lojou, E.; Mazurenko, I. Interplay between Orientation at Electrodes and Copper Activation of Thermus thermophilus Laccase for O2 Reduction. J. Am. Chem. Soc. 2020, 142, 1394–1405, doi:10.1021/jacs.9b11147.
- Tang, J.; Werchmeister, R.M.L.; Preda, L.; Huang, W.; Zheng, Z.; Leimkühler, S.; Wollenberger, U.; Xiao, X.; Engelbrekt, C.; Ulstrup, J.; et al. Three-Dimensional Sulfite Oxidase Bioanodes Based on Graphene Functionalized Carbon Paper for Sulfite/O2 Biofuel Cells. ACS Catal. 2019, 9, 6543–6554, doi:10.1021/acscatal.9b01715.
- Ciaccafava, A.; Infossi, P.; Ilbert, M.; Guiral, M.; Lecomte, S.; Giudici-Orticoni, M.T.; Lojou, E. Electrochemistry, AFM, and PM-IRRA spectroscopy of immobilized hydrogenase: Role of a hydrophobic helix in enzyme orientation for efficient H2 oxidation. Angew. Chem. Int. Ed. 2012, 51, 953–956, doi:10.1002/anie.201107053.
- Loew, N.; Scheller, F.W.; Wollenberger, U. Characterization of self-assembling of glucose dehydrogenase in mono- and multilayers on gold electrodes. Electroanalysis 2004, 16, 1149–1154, doi:10.1002/elan.200403005.
- Yehezkeli, O.; Tel-Vered, R.; Raichlin, S.; Willner, I. Nano-engineered flavin-dependent glucose dehydrogenase/gold nanoparticle-modified electrodes for glucose sensing and biofuel cell applications. ACS Nano 2011, 5, 2385–2391, doi:10.1021/nn200313t.
- Madden, C.; Vaughn, M.D.; Díez-Pérez, I.; Brown, K.A.; King, P.W.; Gust, D.; Moore, A.L.; Moore, T.A. Catalytic turnover of [FeFe]-hydrogenase based on single-molecule imaging. J. Am. Chem. Soc. 2012, 134, 1577–1582, doi:10.1021/ja207461t.
- Hitaishi, V.P.; Mazurenko, I.; Harb, M.; Clément, R.; Taris, M.; Castano, S.; Duché, D.; Lecomte, S.; Ilbert, M.; De Poulpiquet, A.; et al. Electrostatic-Driven Activity, Loading, Dynamics, and Stability of a Redox Enzyme on Functionalized-Gold Electrodes for Bioelectrocatalysis. ACS Catal. 2018, 8, 12004–12014, doi:10.1021/acscatal.8b03443.
- Guo, L.H.; Allen, H.; Hill, O. Direct electrochemistry of proteins and enzymes. Adv. Inorg. Chem. 1991, 36, 341–375, doi:10.1016/S0898-8838(08)60043-4.
- Albery, W.J.; Eddowes, M.J.; Allen, H.; Hill, O.; Hillman, A.R. Mechanism of the Reduction and Oxidation Reaction of Cytochrome c at a Modified Gold Electrode. J. Am. Chem. Soc. 1981, 103, 3904–3910, doi:10.1021/ja00403a049.
- Armstrong, F.A.; Hill, H.A.O.; Walton, N.J. Direct Electrochemistry of Redox Proteins. Acc. Chem. Res. 1988, 21, 407–413, doi:10.1021/ar00155a004.
- Armstrong, F.A.; Heering, H.A.; Hirst, J. Reaction of complex metalloproteins studied by protein-film voltammetry. Chem. Soc. Rev. 1997, 26, 169–179, doi:10.1039/cs9972600169.
- Ma, S.; Laurent, C.V.F.P.; Meneghello, M.; Tuoriniemi, J.; Oostenbrink, C.; Gorton, L.; Bartlett, P.N.; Ludwig, R. Direct electron-transfer anisotropy of a site-specifically immobilized cellobiose dehydrogenase. ACS Catal. 2019, 9, 7607–7615, doi:10.1021/acscatal.9b02014.
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. Highly Sensitive Membraneless Fructose Biosensor Based on Fructose Dehydrogenase Immobilized onto Aryl Thiol Modified Highly Porous Gold Electrode: Characterization and Application in Food Samples. Anal. Chem. 2018, 90, 12131–12136, doi:10.1021/acs.analchem.8b03093.


