 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Florian Perner | + 2749 word(s) | 2749 | 2021-01-29 03:11:24 | | | |
| 2 | Florian Perner | Meta information modification | 2749 | 2021-02-01 01:47:17 | | | | |
| 3 | Peter Tang | Meta information modification | 2749 | 2021-02-01 02:16:50 | | |
Video Upload Options
The aberrant function of chromatin regulatory networks (epigenetics) is a hallmark of cancer promoting oncogenic gene expression. A growing body of evidence suggests that the disruption of specific chromatin-associated protein complexes has therapeutic potential in malignant conditions, particularly those that are driven by aberrant chromatin modifiers. Of note, a number of enzymatic inhibitors that block the catalytic function of histone modifying enzymes have been established and entered clinical trials. Unfortunately, many of these molecules do not have potent single-agent activity. One potential explanation for this phenomenon is the fact that those drugs do not profoundly disrupt the integrity of the aberrant network of multiprotein complexes on chromatin.
1. Introduction
The genomic DNA in eukaryotic cells is wrapped around a core histone octamer composed of Histone H2A (H2A), Histone H2B (H2B), Histone H3 (H3), and Histone H4 (H4) [1][2][3]. This complex of DNA and histones is compacted at different densities to chromatin, ranging from a simple chain of DNA and histones to highly condensed metaphase chromosomes [4][5][6][7][8][9]. The tight regulation of chromatin condensation and de-condensation is vital for a healthy organism since it is crucial for the equal distribution of genetic information to the daughter cells during cell division [10][11]. Furthermore, the state of chromatin condensation and therefore the degree of DNA accessibility, restricts or allows transcription factors and the RNA-polymerase machinery to physically interact with the DNA. Therefore, the stringent regulation of chromatin accessibility is a critical determinant of proper spatial and temporal regulation of gene transcription [12][13][14]. In cancer cells, the healthy chromatin homeostasis is disrupted by a variety of mechanisms promoting aberrant transcription and potentially also cytogenetic alterations [15][16][17].
The dynamics of chromatin biology are regulated by a variety of post-translational modifications at the unstructured core-histone tails [18]. These modifications involve methylation, acetylation, and phosphorylation, as well as many other less well characterized modifications like sumoylation, ADP-ribosylation, deimination, proline-isomerization, and crotonylation [18][19][20][21][22]. Early studies in the field have provided evidence that histone acetylation, a mark that is broadly associated with active chromatin, diminishes the positive charge of histones, thereby loosening the interaction between the histones and the negatively charged phosphate groups on the DNA, resulting in increased DNA accessibility [23][24][25][26][27][28][29]. In 2000, Brian Strahl and David Allis suggested a groundbreaking concept by proposing the model of the "histone code" [22]. Several observations from their and other groups prompted the idea that the complex array of post-translational modifications on histone tails serve as a binding platform for reader proteins and associated multiprotein complexes and thereby orchestrate a complex network of regulatory proteins on chromatin [22][30][31][32][33][34]. This concept is the basis for our current understanding of the regulatory dynamics on chromatin involving writers, erasers, and readers of histone modifications (Figure 1). An example is H3K27 acetylation, which is associated with active enhancers and promotors [18]. The "writers" of this mark are the acetyltransferases p300 and CBP that deposit H3K27ac to activate specific enhancers and promotors [35][36]. Histone-deacetylases (like HDAC1 and 2) serve in an antagonistic fashion as "erasers" to remove the mark and repress these regulatory elements. BRD4, that can associate with H3K27ac nucleosomes via its bromodomain, is a "reader" that gets recruited to active promotors and enhancers through binding to H3K27ac [37][38][39][40]. Importantly, BRD4 associates with the multiprotein-complex of the transcription machinery and is essential for the activation of RNA-polymerase II (RNA-PolII) by the superelongation complex [41]. Therefore, altering the function of readers, writers, or erasers in this system will have dramatic consequences for the homeostasis of gene transcription [40][42][43][44][45][46].
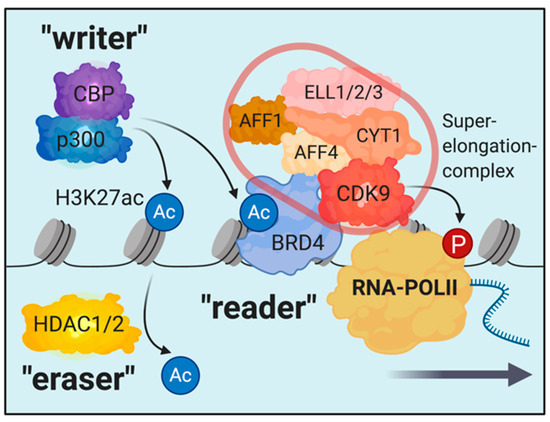
Figure 1. Schematic illustrating the concept of writers, erasers, and readers on the example of the H3K27ac-mark. Multiprotein-complexes are recruited and/or associate with reader proteins for locus specific chromatin localization. In this example, the superelongation-complex, which is required for RNAPolII activation, associates with BRD4.
In actual physiological systems, the complexity of the protein-network generated by the histone code is complicated, since the regulatory elements of different marks are highly interconnected. Writers and erasers of histone modifications get recruited by reader proteins of other histone modifications, leading to a topologically organized array of co-existing modifications and associated multi-protein complexes that define distinct regulatory microenvironments at specific regions on chromatin impacting transcription and 3D-chromosome architecture [47][48][49][50][51][52]. These regulatory networks are central mediators of embryonic development, lineage determination during differentiation and cellular homeostasis [53][54][55][56][57][58][59][60][61][62][63][64][65]. Consequently, aberrant regulation of this network is a hallmark of cancer development and progression [17][66][67][68][69][70]. Several of the recurrently detected genetic abnormalities in patients with myeloid malignancies involve epigenetic modifiers like DNMT3A, TET2, ASXL1, EZH2, IDH1/2, KMT2A, KAT6A, KDM5A, KDM6A, or NSD1 [71][72][73][74]. In the recent years, it has become clear that mutations in some of these genes arise in hematopoietic stem cells of healthy individuals during the process of aging [75][76][77][78]. The occurrence of these mutations was found to be associated with an increased risk to develop myeloid malignancies as well as cardiovascular disease [75][79][80]. The application of novel single-cell sequencing approaches in primary material from patients with acute myeloid leukemia (AML) and myeloproliferative neoplasia (MPN) revealed that these clonal mutations in epigenetic modifiers tend to co-occur in dominant clones of patients that ultimately developed leukemia, implicating a synergistic pathogenic function if occurring in the same cell. Mutations in signaling molecules, on the other hand, appeared to originate later during disease evolution and showed a tendency not to co-occur within the same clone [81]. These recent findings strongly suggest, that epigenetic rewiring in specific hematopoietic clones is the foundation for the development of myeloid malignancies and may explain the increasing incidence of myelodysplastic syndrome (MDS), MPN, and AML in elderly individuals. Nevertheless, no pharmacological targeting approaches that show promise in blocking expansion and molecular progression of these pre-leukemic clones have been established yet clinically. Gaining a better understanding of the chromatin biology during this preleukemic disease state and developing valid predictors of progression, as well as clinically active epigenetic mono- or combination therapies, will be a central goal to prevent leukemia development in the future. However, much progress has been made in the preclinical and clinical development of compounds interfering with the function of chromatin modifiers for use in patients with fully developed cancer. In both myeloid malignancies and solid cancers, several of these molecules have entered clinical trials and show activity in a variety of diseases [74][82][83][84][85].
2. Inhibition of Histone Methylation on Chromatin: Recent Clinical Advances and Challenges
Targeting histone-methylation pattern on chromatin is a scientifically and clinically exiting opportunity to interrogate with disease-related epigenetic programs in vitro and in vivo. Given the fact, that a very specific set of methyltransferases and de-methylases is responsible for the deposition and removal of specific methylation-marks, the interrogation with distinct de-regulated pattern within the histone-code is possible. In contrast, histone acetyltransferases and deacetylases act in a more promiscuous way. Those enzymes have a very broad spectrum of targets among histones as well as non-histone proteins. Therefore, the molecular consequences of inhibiting those enzymes are more complex and much less specific for certain epigenetic programs. Multiple inhibitory drugs for methyltransferases, acetyltransferases, demethylases, and deacetylases have been developed and are currently under preclinical or clinical investigation [83]. In the history of drug-discovery, much experience has been gathered regarding the development and evaluation of enzymatic inhibitors [86][87][88]. Those drugs act as competitive or allosteric inhibitors of catalytically active proteins, preventing the binding of an enzyme to its target substrate and/or disturbing the structural integrity of its catalytic center. In the epigenetic space, proteins with enzymatic activity play a crucial role as "writers" and "erasers" of histone modifications, as discussed above. Therefore, molecules that inhibit the activity of those proteins have the potential to specifically interfere with the global deposition of certain histone-marks. A number of these molecules have been developed for the treatment of hematologic malignancies and solid cancers [83]. The first inhibitors of histone modifiers that have been approved for the treatment of cancer were inhibitors of histone-deacetylases. The first-in class molecule Vorinostat was initially approved for cutaneous T cell lymphoma [89]. Drug development efforts to improve the pharmacologic properties led to the approval of Belinostat in 2014 for T-cell lymphoma and Panobinostat in 2015 for refractory multiple myeloma. In myeloid malignancies, HDAC inhibitors show promise in clinical trials as monotherapy or in combination with other drugs [83][90][91][92]. Using this class of drugs for the precise interrogation of specific epigenetic programs/mechanisms has however remained difficult. As mentioned above, HDACs act in a promiscuous way erasing histone acetylation broadly across the genome. Furthermore, clinically active compounds are mostly pan-HDAC-inhibitors without particular selectivity for a specific molecule. Therefore, distinct oncogenic signatures or somatic mutations that confer sensitivity to HDAC-inhibition have not been established. More recently, the evolving knowledge about epigenetic mechanisms and subsequent drug development efforts have led to the establishment of a number of inhibitors of histone-methyltransferases. Histone methyltransferases appear to act in a more selective fashion than acetyltransferases. This class of compounds may enable scientists and clinicians for the first time to develop mechanistically driven and relatively well tolerated treatment regimens for cancer therapy. In this review, we will focus on specific inhibitors of histone-modifying complexes that have recently entered clinical trials (Table 1).
Table 1. Clinical development stage of histone methyltransferase enzymatic inhibitors.
|
Drug |
Target |
Clinical phase |
Indication |
|
Tazemetostat (EPZ-6438) |
EZH2 |
approved |
Rhabdoid tumors, Follicular lymphoma |
|
Tazemetostat (EPZ-6438) |
EZH2 |
phase 2 |
Diffuse Large B-Cell Lymphoma, Prostate Cancer, Synovial Sarcoma, Epitheloid Sarcoma, Mesothelioma, Squamous-cell Carcinoma, Urothelial Carcinoma, Ovarian and Endometrial Carcinoma, Melanoma, |
|
GSK2816126 |
EZH2 |
phase 1 |
Diffuse Large B Cell Lymphoma, Follicular Lymphoma, Other Non-Hodgkin's Lymphomas, Solid Tumors, Multiple Myeloma |
|
CPI-1202 |
EZH2 |
phase 2 |
Diffuse Large B-Cell Lymphoma, Prostate Cancer, Advanced Solid Tumors |
|
Pinometostat (EPZ-5676) |
DOT1L |
phase 2 |
Acute Myeloid Leukemia, Acute Lymphatic Leukemia |
|
Tranylcypromine |
LSD1 |
phase 2 |
Acute Myeloid Leukemia, Myelodysplastic Syndrome |
|
Iadademstat (ORY-2001) |
LSD1 |
phase 2 |
Alzheimer’s Disease |
|
Bomedemstat (IMG-7289) |
LSD1 |
phase 2 |
Essential Thrombocythemia, Polycythemia vera, Myelofibrosis, Acute Myeloid Leukemia, Myelodysplastic Syndrome |
Summary of drugs discussed in this manuscript with their target, clinical stage, and indications.
3. Disrupting Protein-Protein Interactions to Target Epigenetic Complexes
Despite the recent advances in the development and clinical establishment of enzymatic inhibitors of chromatin modifiers, major challenges remain on the way to development of more clinically efficacious drugs that target chromatin complexes. As described above, most enzymatic inhibitors of histone-modifying enzymes that have been assessed to date show only modest clinical activity, particularly when administrated as single agents. Several of these compounds showed sufficient on-target activity in vivo (as measured by global reduction of histone methylation) and were relatively well tolerated. Still, the clinical responses observed were not as extensive as might have been predicted based on the phenotypes described in preclinical model systems using genetic-inactivation and small molecule inhibitors. In recent years, a growing body of evidence suggests that inhibition of any single enzymatic activity/function in a chromatin complex may be insufficient to perturb epigenetic homeostasis and control gene expression in cancers cells. This has recently been demonstrated for complexes containing EZH2 and LSD1 [93][94][95]. Similarly, pharmacologic inhibition of DOT1L’s methyltransferase function shows modest and delayed responses in vitro and in vivo when compared to genetic inactivation that demonstrated DOT1L’s essential function in MLL-rearranged leukemia [96][97][98][99][100][101][102][103]. Therefore, we expect approaches that disrupt chromatin associated complexes more extensively may be more therapeutically efficacious.
Another major issue is that only a relatively small fraction of essential proteins on chromatin have enzymatic activity, so the spectrum of potential applications for catalytic inhibitors is naturally limited. Recent advances in the development of inhibitors of protein–protein interactions open up a novel field of potential applications, particularly in the epigenetic space, were a large proportion of the dense network of multiprotein-complexes cannot be pharmacologically addressed using enzymatic inhibitors. In the past few years, a variety of compounds disrupting epigenetic protein–protein interactions have been synthesized and established mostly in vitro [104]. In this section, we will focus on Menin-MLL inhibitors and bromodomain-inhibitors since these molecules have already been intensively investigated in preclinical studies and entered early-phase clinical trials.
4. Hijacking the Proteasome for Targeted Protein Degradation
In the recent years, targeted protein degradation has evolved as a novel and emerging strategy in drug development [105][106][107]. Two major concepts to achieve targeted protein degradation have been established and are currently exploited in drug discovery:
"Immunomodulatory drugs" like the FDA-approved compounds Thalidomide, Lenalidomide, and Pomalidomide exert their therapeutic function by selective degradation of a variety of zinc-finger transcription factors, including IKZF1 and IKZF3. Those molecules act as a "molecular glue" that binds to its target and subsequently recruits a cereblon E3-ligase complex, leading to polyubiquitination and proteasomal degradation of the target [108][109]. Recent advances in drug discovery strategies have led to screening approaches that allow to exploit this molecular glue concept to design degraders that specifically target other target proteins, opening up the opportunity to address a variety of drug targets including epigenetic modifiers in the future [110][111].
PROteolysis TArgeting Chimeras (PROTACs) are bifunctional molecules high affinity ligands of ubiquitin ligases like cereblon (CRBN), Von Hippel-Lindau (VHL), or DCAF16 are linked to a highly specific binder to the protein of interest [106][107]. The design and application of PROTACs is rapidly evolving in part due to the fact that highly selective binders of target protein are available for a variety of target proteins in the form of established small molecules that bind to enzymatic pockets or other protein domains. PROTACs successfully targeting kinases, nuclear receptors, and epigenetic modulators have recently been established [106].
In the space of bromodomain-targeting, the differences between bromodomain-inhibition and targeted bromodomain-protein-degradation have been demonstrated and exemplify the potential of this novel targeting approach. In contrast to BET-competitive inhibition, targeted degradation of BRD4 using the optimized degrader molecule dBET6 led to a global breakdown of transcription, therefore having more rapid and dramatic consequences in cancer cells [112]. The partial selectivity of BRD4-inhibtion for cancer specific enhancer and super-enhancer programs was not observed with this BRD4 degrader. This data demonstrated, that indeed BRD4 is a common essential gene with a non-redundant function in transcriptional elongation in malignant as well as normal cells. Therefore, the degradation of BRD4 has detrimental consequences, indicating a clear mechanistic difference to bromodomain-inhibition. Presumably, these differences are due to a more rapid and profound disruption of the transcription machinery at actively transcribed genes. Even though it might be challenging to manage toxicity of this type of treatment in more advanced pre-clinical stages of development, BET-degradation by dBET6 has been shown to be an efficient and tolerable treatment strategy in a mouse model of glioblastoma, indicating a potential therapeutic window in some transcriptionally driven cancers [113]. Nevertheless, based on the current data, it is questionable if BRD4 degradation is a viable therapeutic approach for targeted therapy rather than a cytotoxic treatment with a therapeutic window in aggressive cancers, similar to chemotherapy. However, these data clearly demonstrate different functional consequences when one uses small molecules to inhibit a protein’s function as compared to complete eradication of the protein.
A clearer example for the potential therapeutic advantages of bromodomain-protein degradation are the pre-clinical observations made in synovial sarcoma regarding targeting of BRD9. As opposed to BRD4, BRD9 is a selective dependency in certain cancer types, including AML and synovial sarcoma [114][115][116]. BRD9 is a part of the mammalian SWI/SNF chromatin remodeling complex, more specifically the ncBAF complex [117][118]. Dysfunctional BAF-complexes have a specific relevance in synovial sarcoma, since the disease is driven by SS18-SSX fusion oncogenes. SS18 itself is a part of mammalian BAF-complexes, therefore, its involvement in oncogenic fusions in synovial sarcoma drives aberrant SWI/SNF function. Importantly, genetic targeting of BRD9 using different single-guide RNAs was detrimental for synovial sarcoma cells. A selective inhibitor of the BRD9-bromodomain on the other hand showed only modest cellular activity at reasonable drug concentrations. ChIP-sequencing demonstrated that BRD9-bromodomain inhibition only led to a sub-complete dissociation of BRD9 from chromatin (Figure 2). When utilizing a PROTAC of BRD9 (dBRD9A), the therapeutic efficacy in synovial sarcoma could be substantially improved due to elimination of BRD9 and a suspected higher degree of ncBAF complex disruption [114]. These findings have prompted the development of orally bioavailable degraders of BRD9. The first clinical trial investigating one of these compounds in synovial sarcoma are projected to start by the end of 2021.
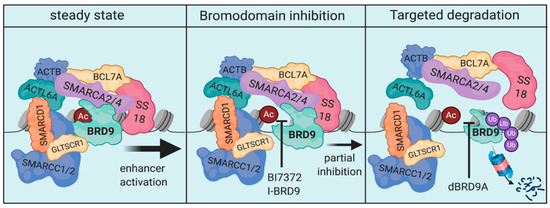
Figure 2. Schematic of the ncBAF-complex and the molecular consequences of BRD9-inhibition and BRD9-degradation. The chromatin remodeling function of the ncBAF complex is essential in maintenance of SS18-fusion driven synovial sarcoma (left). BRD9-bromodomain-inhibition leads to only a partial dissociation of the complex limiting cellular efficacy (middle). Targeted degradation of BRD9, on the other hand, de-stabilizes the complex to a greater extent, enabling increased cellular efficacy (right).
References
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150, doi:10.1038/nature01595.
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260, doi:10.1038/38444.
- Richmond, T.J.; Finch, J.T.; Rushton, B.; Rhodes, D.; Klug, A. Structure of the nucleosome core particle at 7 A resolution. Nature 1984, 311, 532–537, doi:10.1038/311532a0.
- Nicodemi, M.; Pombo, A. Models of chromosome structure. Curr. Opin. Cell Biol. 2014, 28, 90–95, doi:10.1016/j.ceb.2014.04.004.
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871, doi:10.1126/science.184.4139.868.
- Olins, A.L.; Olins, D.E. Spheroid chromatin units (v bodies). Science 1974, 183, 330–332, doi:10.1126/science.183.4122.330.
- Widom, J.; Klug, A. Structure of the 300A chromatin filament: X-ray diffraction from oriented samples. Cell 1985, 43, 207–213, doi:10.1016/0092-8674(85)90025-x.
- Belmont, A.S.; Bruce, K. Visualization of G1 chromosomes: A folded, twisted, supercoiled chromonema model of interphase chromatid structure. J. Cell Biol. 1994, 127, 287–302, doi:10.1083/jcb.127.2.287.
- Gerchman, S.E.; Ramakrishnan, V. Chromatin higher-order structure studied by neutron scattering and scanning transmission electron microscopy. Proc. Natl. Acad. Sci. USA 1987, 84, 7802–7806, doi:10.1073/pnas.84.22.7802.
- Vagnarelli, P. Chapter Six—Chromatin Reorganization Through Mitosis. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Boston, MA, USA, 2013; Volume 90, pp. 179–224.
- Antonin, W.; Neumann, H. Chromosome condensation and decondensation during mitosis. Curr. Opin. Cell Biol. 2016, 40, 15–22, doi:10.1016/j.ceb.2016.01.013.
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719, doi:10.1016/j.cell.2007.01.015.
- Voss, T.C.; Hager, G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014, 15, 69–81, doi:10.1038/nrg3623.
- Rando, O.J.; Winston, F. Chromatin and transcription in yeast. Genetics 2012, 190, 351–387, doi:10.1534/genetics.111.132266.
- Valencia, A.M.; Kadoch, C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 2019, 21, 152–161, doi:10.1038/s41556-018-0258-1.
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249, doi:10.1101/gad.255182.114.
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469, doi:10.1038/nrc2876.
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705, doi:10.1016/j.cell.2007.02.005.
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324, doi:10.1038/emm.2017.11.
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903, doi:10.1038/ng.154.
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028, doi:10.1016/j.cell.2011.08.008.
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45, doi:10.1038/47412.
- Wolffe, A.P.; Hayes, J.J. Chromatin disruption and modification. Nucleic Acids Res. 1999, 27, 711–720, doi:10.1093/nar/27.3.711.
- Bauer, W.R.; Hayes, J.J.; White, J.H.; Wolffe, A.P. Nucleosome structural changes due to acetylation. J. Mol. Biol. 1994, 236, 685–690, doi:10.1006/jmbi.1994.1180.
- Krajewski, W.A.; Becker, P.B. Reconstitution of hyperacetylated, DNase I-sensitive chromatin characterized by high conformational flexibility of nucleosomal DNA. Proc. Natl. Acad. Sci. USA 1998, 95, 1540–1545, doi:10.1073/pnas.95.4.1540.
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626, doi:10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H.
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J. Biol. Chem. 1993, 268, 305–314.
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851, doi:10.1016/s0092-8674(00)81063-6.
- Durrin, L.K.; Mann, R.K.; Kayne, P.S.; Grunstein, M. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell 1991, 65, 1023–1031, doi:10.1016/0092-8674(91)90554-c.
- Turner, B.M. Decoding the nucleosome. Cell 1993, 75, 5–8.
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352, doi:10.1038/38664.
- Mizzen, C.; Kuo, M.H.; Smith, E.; Brownell, J.; Zhou, J.; Ohba, R.; Wei, Y.; Monaco, L.; Sassone-Corsi, P.; Allis, C.D. Signaling to chromatin through histone modifications: How clear is the signal? Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 469–481, doi:10.1101/sqb.1998.63.469.
- Lopez-Rodas, G.; Brosch, G.; Georgieva, E.I.; Sendra, R.; Franco, L.; Loidl, P. Histone deacetylase. A key enzyme for the binding of regulatory proteins to chromatin. FEBS Lett. 1993, 317, 175–180, doi:10.1016/0014-5793(93)81271-z.
- Tordera, V.; Sendra, R.; Perez-Ortin, J.E. The role of histones and their modifications in the informative content of chromatin. Experientia 1993, 49, 780–788, doi:10.1007/BF01923548.
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452, doi:10.1021/cr500452k.
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373.
- Brown, J.D.; Feldman, Z.B.; Doherty, S.P.; Reyes, J.M.; Rahl, P.B.; Lin, C.Y.; Sheng, Q.; Duan, Q.; Federation, A.J.; Kung, A.L.; et al. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 2144–2149, doi:10.1073/pnas.1711155115.
- Lee, J.E.; Park, Y.K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 2217, doi:10.1038/s41467-017-02403-5.
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790, doi:10.1016/j.ccr.2013.11.003.
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334, doi:10.1016/j.cell.2013.03.036.
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545, doi:10.1016/j.molcel.2005.06.029.
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. 2014, 8, 1919–1929, doi:10.1016/j.celrep.2014.08.025.
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528, doi:10.1038/nature10334.
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073, doi:10.1038/nature09504.
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729, doi:10.1016/j.celrep.2018.07.041.
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132, doi:10.1038/nature24028.
- Wilson, S.; Filipp, F.V. A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer. NPJ Syst. Biol. Appl. 2018, 4, 24, doi:10.1038/s41540-018-0061-4.
- Wang, Y.; Jiang, R.; Wong, W.H. Modeling the causal regulatory network by integrating chromatin accessibility and transcriptome data. Natl. Sci. Rev. 2016, 3, 240–251, doi:10.1093/nsr/nww025.
- Tu, X.; Mejia-Guerra, M.K.; Valdes Franco, J.A.; Tzeng, D.; Chu, P.Y.; Shen, W.; Wei, Y.; Dai, X.; Li, P.; Buckler, E.S.; et al. Reconstructing the maize leaf regulatory network using ChIP-seq data of 104 transcription factors. Nat. Commun. 2020, 11, 5089, doi:10.1038/s41467-020-18832-8.
- Malysheva, V.; Mendoza-Parra, M.A.; Blum, M.; Spivakov, M.; Gronemeyer, H. Gene regulatory network reconstruction incorporating 3D chromosomal architecture reveals key transcription factors and DNA elements driving neural lineage commitment. bioRxiv 2019, 303842, doi:10.1101/303842.
- Ashoor, H.; Chen, X.; Rosikiewicz, W.; Wang, J.; Cheng, A.; Wang, P.; Ruan, Y.; Li, S. Graph embedding and unsupervised learning predict genomic sub-compartments from HiC chromatin interaction data. Nat. Commun. 2020, 11, 1173, doi:10.1038/s41467-020-14974-x.
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200, doi:10.1016/j.cell.2016.10.024.
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658, doi:10.1038/nrg.2017.57.
- Amit, I.; Winter, D.R.; Jung, S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat. Immunol. 2016, 17, 18–25, doi:10.1038/ni.3325.
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780, doi:10.1126/science.aad9780.
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484, doi:10.1038/nature08911.
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57, doi:10.1016/j.cell.2017.08.002.
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187, doi:10.1016/j.cell.2016.09.018.
- Papp, B.; Plath, K. Epigenetics of reprogramming to induced pluripotency. Cell 2013, 152, 1324–1343, doi:10.1016/j.cell.2013.02.043.
- Surani, M.A.; Hayashi, K.; Hajkova, P. Genetic and epigenetic regulators of pluripotency. Cell 2007, 128, 747–762, doi:10.1016/j.cell.2007.02.010.
- Yadav, T.; Quivy, J.P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332–1336, doi:10.1126/science.aat8950.
- Apostolou, E.; Hochedlinger, K. Chromatin dynamics during cellular reprogramming. Nature 2013, 502, 462–471, doi:10.1038/nature12749.
- Johnson, D.G.; Dent, S.Y. Chromatin: Receiver and quarterback for cellular signals. Cell 2013, 152, 685–689, doi:10.1016/j.cell.2013.01.017.
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839, doi:10.1016/j.cell.2016.07.050.
- Dulac, C. Brain function and chromatin plasticity. Nature 2010, 465, 728–735, doi:10.1038/nature09231.
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299, doi:10.1038/nrg.2016.13.
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707, doi:10.1038/nrc.2016.82.
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, doi:10.1126/science.aal2380.
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152, doi:10.1126/science.aam7304.
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570, doi:10.1126/science.1230184.
- Fathi, A.T.; Abdel-Wahab, O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv. Hematol. 2012, 2012, 469592, doi:10.1155/2012/469592.
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612, doi:10.1038/nrc3343.
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2015, 263, 22–35, doi:10.1111/imr.12246.
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52, doi:10.1182/blood-2015-07-604512.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498, doi:10.1056/NEJMoa1408617.
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333, doi:10.1038/nature13038.
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657, doi:10.1007/s12185-013-1407-8.
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282, doi:10.1038/leu.2014.211.
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, doi:10.1126/science.aan4673.
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121, doi:10.1056/NEJMoa1701719.
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, doi:10.1038/s41586-020-2864-x.
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207, doi:10.1146/annurev-pharmtox-010716-105106.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62, doi:10.1038/s41392-019-0095-0.
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174, doi:10.1186/s13148-019-0776-0.
- Park, J.W.; Han, J.W. Targeting epigenetics for cancer therapy. Arch. Pharm. Res. 2019, 42, 159–170, doi:10.1007/s12272-019-01126-z.
- Holdgate, G.A.; Meek, T.D.; Grimley, R.L. Mechanistic enzymology in drug discovery: A fresh perspective. Nat. Rev. Drug Discov. 2018, 17, 115–132, doi:10.1038/nrd.2017.219.
- Copeland, R.A.; Harpel, M.R.; Tummino, P.J. Targeting enzyme inhibitors in drug discovery. Expert Opin. Ther. Targets 2007, 11, 967–978, doi:10.1517/14728222.11.7.967.
- Silverman, R.B.; Holladay, M.W. Chapter 5—Enzyme Inhibition and Inactivation. In The Organic Chemistry of Drug Design and Drug Action, 3rd ed.; Silverman, R.B., Holladay, M.W., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 207–274.
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90, doi:10.1038/nbt1272.
- Bose, P.; Verstovsek, S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin. Investig. Drugs 2016, 25, 1393–1403, doi:10.1080/13543784.2016.1250882.
- San Jose-Eneriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers (Basel) 2019, 11, doi:10.3390/cancers11111794.
- Quintas-Cardama, A.; Santos, F.P.; Garcia-Manero, G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235, doi:10.1038/leu.2010.276.
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820 e2804, doi:10.1016/j.celrep.2018.11.035.
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496, doi:10.1038/nm.3968.
- Ravasio, R.; Ceccacci, E.; Nicosia, L.; Hosseini, A.; Rossi, P.L.; Barozzi, I.; Fornasari, L.; Zuffo, R.D.; Valente, S.; Fioravanti, R.; et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci. Adv. 2020, 6, eaax2746, doi:10.1126/sciadv.aax2746.
- Nguyen, A.T.; He, J.; Taranova, O.; Zhang, Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011, 21, 1370–1373, doi:10.1038/cr.2011.115.
- Bernt, K.M.; Armstrong, S.A. A role for DOT1L in MLL-rearranged leukemias. Epigenomics 2011, 3, 667–670, doi:10.2217/epi.11.98.
- Chen, C.W.; Koche, R.P.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.G.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343, doi:10.1038/nm.3832.
- Perner, F.; Gadrey, J.Y.; Xiong, Y.; Hatton, C.; Eschle, B.K.; Weiss, A.; Stauffer, F.; Gaul, C.; Tiedt, R.; Perry, J.A.; et al. Novel Inhibitors of the Histone-Methyltransferase DOT1L Show Potent Antileukemic Activity in Patient-derived Xenografts. Blood 2020, doi:10.1182/blood.2020006113.
- Chan, A.K.N.; Chen, C.W. Rewiring the Epigenetic Networks in MLL-Rearranged Leukemias: Epigenetic Dysregulation and Pharmacological Interventions. Front. Cell Dev. Biol. 2019, 7, 81, doi:10.3389/fcell.2019.00081.
- Okuda, H.; Stanojevic, B.; Kanai, A.; Kawamura, T.; Takahashi, S.; Matsui, H.; Takaori-Kondo, A.; Yokoyama, A. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Investig. 2017, 127, 1918–1931, doi:10.1172/JCI91406.
- Chen, C.W.; Armstrong, S.A. Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol. 2015, 43, 673–684, doi:10.1016/j.exphem.2015.05.012.
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541, doi:10.1182/blood-2012-11-465120.
- Linhares, B.M.; Grembecka, J.; Cierpicki, T. Targeting epigenetic protein-protein interactions with small-molecule inhibitors. Future Med. Chem. 2020, 12, 1305–1326, doi:10.4155/fmc-2020-0082.
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944, doi:10.1038/s41589-019-0362-y.
- Wu, T.; Yoon, H.; Xiong, Y.; Dixon-Clarke, S.E.; Nowak, R.P.; Fischer, E.S. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat. Struct. Mol. Biol. 2020, 27, 605–614, doi:10.1038/s41594-020-0438-0.
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963, doi:10.1038/s41573-019-0047-y.
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305, doi:10.1126/science.1244851.
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309, doi:10.1126/science.1244917.
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207, doi:10.1038/s41589-020-0594-x.
- Slabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297, doi:10.1038/s41586-020-2374-x.
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18.e19, doi:10.1016/j.molcel.2017.06.004.
- Xu, L.; Chen, Y.; Mayakonda, A.; Koh, L.; Chong, Y.K.; Buckley, D.L.; Sandanaraj, E.; Lim, S.W.; Lin, R.Y.; Ke, X.Y.; et al. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. USA 2018, 115, E5086–5095, doi:10.1073/pnas.1712363115.
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 2018, 7, doi:10.7554/eLife.41305.
- Hohmann, A.F.; Martin, L.J.; Minder, J.L.; Roe, J.S.; Shi, J.; Steurer, S.; Bader, G.; McConnell, D.; Pearson, M.; Gerstberger, T.; et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016, 12, 672–679, doi:10.1038/nchembio.2115.
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420, doi:10.1038/s41556-018-0221-1.
- Mashtalir, N.; Suzuki, H.; Farrell, D.P.; Sankar, A.; Luo, J.; Filipovski, M.; D’Avino, A.R.; St Pierre, R.; Valencia, A.M.; Onikubo, T.; et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 2020, 183, 802–817.e824, doi:10.1016/j.cell.2020.09.051.
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e1220, doi:10.1016/j.cell.2018.09.032.

