 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Javier Molina-Cerrillo | + 2322 word(s) | 2322 | 2021-01-29 07:58:49 | | | |
| 2 | Camila Xu | Meta information modification | 2322 | 2021-02-02 05:23:32 | | |
Video Upload Options
Fibroblast growth factors (FGFs) are a family of 22 cell-signaling proteins of extracellular origin, generally released upon tissue injury, which act as systemic or locally circulating molecules capable of activating tyrosine-kinase receptors.
1. Molecular Biology of Fibroblast Growth Factor Receptor (FGFR)
Fibroblast growth factors (FGFs) have been classified in seven subfamilies according to their phylogeny: five paracrine FGFs (FGF1, FGF4, FGF7, FGF9 and FGF8), an endocrine FGF (FGF15/19) and an intracellular subgroup (FGF11). These receptors have a beta-trefoil fold with a heparan sulfate binding-site that facilitates its sequestration close to the cell surface for binding to an FGF receptor (FGFR) [1].
FGFRs are encoded by four different genes (FGFR1–FGFR4) and are composed of three extracellular immunoglobulin-type domains (D1, D2 and D3), with D3 mediating heparan-sulfate binding and being primarily responsible for ligand specificity. The dimerization of the FGFR intracellular-domain precedes an autophosphorylation signal for the tyrosine-kinase domain that leads to the activation of several downstream transduction pathways [2].
Mainly, two different mechanisms have been described in the further transmission of the signal. The first one is the activation of RAS-dependent mitogen activated protein-kinase (MAPK) and Raf phosphorylation. The second one leads to cell activation through other signaling molecules, such as Shb, Src kinase and STATs (signal transducers and activators of transcription), amongst others. The whole FGF/FGFR pathway is strongly regulated by feedback mechanisms, such us SPRY (which down-regulates the activation of growth factor receptor-bound protein) and MKP3 (which attenuates MAPK signaling) [3] (Figure 2).
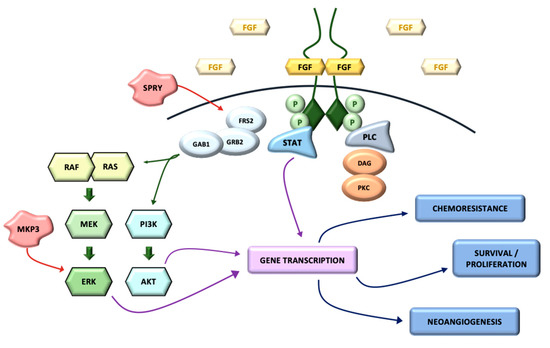
Figure 2. FGFR signaling pathway.
In non-cancer cells, the activation of FGFRs leads to the stimulation of several intracellular signaling cascades that play crucial roles in embryonic development, metabolism and tissue repair. Due to the significant influences of FGF/FGFR pathway on cell growth, proliferation and differentiation, its dysregulation secondarily to different kinds of genetic aberrations (including receptor mutations, amplifications and chromosomal translocations) has an important oncogenic role, especially related to tumor progression and resistance to CT. Around 7.1% of all tumor types present genetic alterations in the FGF/FGFR axis, FGFR1 being the most frequently altered (49%), followed by FGFR3 and FGFR2—hence it is the third most frequently altered pathway after TP53 and KRAS [4].
Specifically, amplifications of FGFR1 gene have been found in 9–10% of urothelial BC, followed by FGFR3 (3–5%) and FGFR2 (0.8%), and activating mutations of FGFR3 gene have been described in 38–66% of non-invasive BC and 15–20% of invasive BC. Interestingly, for therapeutic purposes, the presence of any FGFR mutation, fusion or overexpression seems to be associated with a higher sensitivity to FGFR inhibitors in pre-clinical models [5].
Amplification of FGFR represents around 66% of FGFR alterations, with FGFR1 being the most frequently amplified subtype. FGFR1 amplification seems to be much more represented in early than advanced-stage tumors, suggesting a possible role of FGFR1 amplification during the initial phase of oncogenesis, which may be clinically relevant for therapeutic purposes [6].
Missense mutations such as FGFR3S249C (21%), FGFR3Y375C (7%), FGFR3R248C (3%) and FGFR3-TACC3 fusions (2%) are relatively common in NMIBC (20–50%) and not rare in MIBC (10%) [7], and they have been related to the aberrant formation of cys-mediated intermolecular bonds between mutant receptors and to the constitutive activation of the FGFR3 tyrosine-kinase [8][9].
Despite these genetic alterations having set the stage for the development of targeted therapies, the modest response rates observed in clinical trials, and the accumulating evidence related to other TKIs, suggest that primary or acquired resistance is an unavoidable concern related to the current FGFR inhibitors. The bypass activation of the same or similar downstream effectors is a known mechanism of both intrinsic and acquired resistance. For example, the activation of EGFR/HER3-dependent PI3K/Akt signaling has been described in urothelial tumors harboring driver FGFR3 mutations such as FGFR3S249C and FGFR3-TACC3, which are intrinsically resistant to FGFR3 inhibition, suggesting that EGFR-dependent PI3K signaling is a potential mechanism of resistance to FGFR inhibitors [10]. A second major cause of resistance to FGFR-targeted therapies is the emergence of secondary FGFR alterations. Gatekeeper mutations, including FGFR1V561M, FGFR2V564F/I, FGFR3V555M and FGFR4V550E/L, can either occur de novo or during treatment with targeted therapies, leading to amino acid substitutions for the valine residue located in the drug-binding pocket of the tyrosine-kinase domain that may alter the mode of drug-FGFR interactions [11]. Intratumor heterogeneity has been also considered involved in the antitumor responses to FGFR targeted therapies. The homogeneous overexpression of FGFR has been shown to confer malignant cells a high sensitivity to FGFR inhibitors, whereas a heterogeneous FGFR upregulation might entail the existence of resistant cell clones.
Further research is necessary to adequately monitor and identify the emergence of resistant tumor subclones with an activation of parallel pathways or secondary FGFR mutations, enabling the detection of treatment resistance and the stratification of patients to receive appropriate targeted therapies.
3.2. Clinical Trials with FGFR inhibitors in urothelial carcinoma
Several compounds have been developed in recent years to inhibit FGFR. Some of them are non-selective multi-target inhibitors, and others are highly selective FGFR-TKIs, although other approaches, such as monoclonal antibodies and FGF-ligand traps, are also under research. Table 2 shows the more relevant clinical trials targeting FGFR.
Table 2. Clinical trials of FGFR inhibitors.
| Study Design (NCT Identifier and Code) | Study Phase | Experimental Treatment | Population | Estimated n | Primary Endpoint | Estimated Study Completion Date |
|---|---|---|---|---|---|---|
| BLC2001 (NCT02365597) | Phase II | Erdafitinib | mUC with FGR3 mutation or FGFR2/3 fusion afterchemotherapy treatment | 236 | ORR | 30 June 2022 (Recruiting) |
| NCT03390504 | Phase III | Erdafitinib Pembrolizumab |
mUC with FGFR alterations as second or third line of treatment | 631 | OS | 5 November 2021 (Recruiting) |
| NORSE study (NCT03473743) | Phase I/II | Erdafitinib+cetrelimimab Erdafitinib+(cis/carbo)platin |
mUC with selected FGFR alterations | 160 | DLT | 17 March 2023 (Recruiting) |
| NCT04172675 | Phase II | Erdafitinib | NMIBC with FGFR mutations or fusions and recurred after BCG therapy | 280 | RFS | 10 June 2026 (Recruiting) |
| NCT01004224 | Phase I | Infigratinib | Solid tumors with FGFR alterations | 208 | DLP | 8 October 2018 (Completed) |
| NCT04197986 | Phase III | Infigratinib | UC with FGFR3 alterations as adjuvant treatment | 218 | OS | 31 January 2025 (Recruiting) |
| NCT01976741 | Phase I | Rogaratinib | Several solid tumors without/with FGFR alterations | 168 | DLP | 11 March 2019 (Completed) |
| FORT-1 (NCT03410693) | Phase II/III | Rogaratinib | mUC with FGFR1/3 after platinum-based chemotherapy | 172 | ORR | 27 October 2020 (Completed) |
| FORT-2 (NCT03473756) | Phase Ib/II | Rogaratinib+atezolizumab | UC with FGFR alterations as first line of treatment | 210 | DLP | 4 September 2024 (Recruiting) |
| FIGHT-201 (NCT02872714) | Phase II | Pemigatinib | mUC with FGFR alterations | 263 | ORR | 31 March 2021 (Active, no recruiting) |
| FIGHT-205 (NCT04003610) | Phase II | Pemigatinib+atezolizumab Pemigatinib |
mUC with FGFR3 alteration and not eligible to cisplatin | 6 | PFS | 31 January 2026 (Recruiting) |
| NCT02052778 | Phase I | TAS 120 | Tumors with FGF/FGFR alterations | 386 | DLT | 29 May 2021 (Active, not recruiting) |
| NCT01948297 | Phase I | Debio 1347-101 | Tumors with FGFR 1, 2, 3 alterations | 77 | DLT | 26 June 2020 (Terminated) |
| BISCAY (NCT02546661) | Phase I | AZD4547 AZD4547+durvalumab |
MIBC who progressed prior line of treatment | 156 | DLT | 14 February 2022 (Active, not recruiting) |
| NCT04045613 | Phase I/II | Derazantinib Atezolizumab Derazantinib ± atezolizumab |
mUC with FGFR alterations | 306 | ORR | Recruiting (May 2022) |
| NCT00790426 | Phase II | Dovitinib | UC | 48 | OS | April 2012 (Completed) |
| NCT01732107 | Phase II | Dovitinib | NMIUC with FGFR3 alterations | 13 | ORR | 6 March 2017 (Completed) |
Erdafitinib is a novel pan-FGFR kinase inhibitor recently approved by the FDA for patients with locally advanced cancer or mUC with susceptible FGFR3 or FGFR2 genetic alterations who have progressed during or following platinum-based CT [12]. Approval was based on data from the primary analysis of the BLC2001 study [13]. The final results of this phase II trial were presented at ASCO 2020, including long-term outcomes and safety data. With a median follow-up of 24 months, the investigators confirmed an ORR of 40%, with a median duration of response of 6 months. Furthermore, 31% of responders had a duration of response over 12 months. mPFS was 5.52 months and mOS was 11.3 months. Central serous retinopathy (CSR) occurred in 27% (27/101) of patients, but 85% of those (23/27) were grade 1 or 2 [14]. In addition, a phase III trial is evaluating erdafitinib compared to pembrolizumab or CT in patients with mUC and FGFR alterations who have progressed after one or two prior treatments (NCT03390504) [15].
Furthermore, the combination of FGFR inhibition and IT has been analyzed with different agents. The rationale for this strategy is based on different hypothesis. IT may enhance the antitumor effects of FGFR inhibitors and also prevent or delay the development of resistance. Urothelial carcinoma can be divided into T-cell-inflamed and non-T-cell-inflamed subtypes [16]. Non-T-cell-inflamed subtypes correlated with an absence of CD8+ T lymphocyte and resistance to IT, which produced a rationale for a combination of FGFR inhibitors and anti-PD-1/PD-L1 [17]. The aim of the combination of an FGFR inhibitor and an anti-PD-1/PD-L1, such as NORSE study, FORT-2 or FIGHT-205, is to prove that targeting FGFR makes it possible to turn an immunologically cold tumor into a hot tumor.
Therefore, a phase Ib/II clinical trial (NORSE study) evaluated erdafitinib in combination with cetrelimab, a PD-1 inhibitor, in 15 patients with mUC and FGFR2/3 alterations after progression to at least one line of treatment. The combination of erdafitinib (8 mg with uptitration to 9 mg) with cetrelimab was deemed safe for further evaluation [18]. In the seven patients treated with the recommended phase II dose, ORR was 71%. This combination is further being evaluated in a randomized phase II clinical trial in combination with platin-based CT (NCT03473743). However, in high risk, BCG refractory NMIBC with FGFR gene alterations, erdafitinib is being compared with intravesical CT (NCT 04172675).
Infigratinib (BGJ398) is an oral, selective, ATP-competitive FGFR 1–3 TKI. A phase I clinical trial evaluated the safety and antitumor activity of infigratinib in 132 patients with solid tumors [19]. Thirty-three mUC patients with activating FGFR3 mutations or fusions received BGJ398 125 mg on a once-a-day, 3 weeks on/1 week off regimen. Median treatment duration was 13.3 weeks. ORR was 35% [20]. This drug is under development in other UC settings, such as in the perioperative context and in upper urothelial tract (a promising response has been identified in a phase I trial [21]). A phase III clinical trial is currently evaluating infigratinib in patients with UC in the bladder and upper tract in the adjuvant setting (NCT04197986) [22].
Rogaratinib is an oral and selective FGFRs 1–4 TKI that inhibits the auto-phosphorylation of FGFR. A phase I trial tested rogaritinib in patients with advanced solid tumors who were FGFR mRNA-positive. In the mUC cohort, the ORR was 20.8%, with one patient achieving a complete response, and the disease control rate (DCR) was 68.1% [23].
The FORT-1 study evaluated the efficacy of rogaratinib in comparison with CT in patients with mUC who received prior platin-based CT [24]. Patients were included according to FGFR 1 and 3 mRNA expression, analyzed centrally by in situ hybridization from archival tumor tissue; 175 patients were randomized in the study—87 to the rogaratinib arm and 88 to the chemotherapy arm. The ORRs were 19.5% and 19.3% (1-sided p = 0.56), and mPFS values were 2.7 (95% CI, 1.6–4.2) vs. 2.9 (95% CI, 2.6–4.2) months for rogaratinib and CT, respectively. In the exploratory analysis directed at patients with FGFR3 DNA mutations or fusions, ORR was 52.4% for rogaratinib—higher compared to CT’s 26.7%. Considering these results, the study terminated early.
FORT-2 is a phase Ib/II study that evaluates the safety and efficacy of rogaratinib in combination with atezolizumab, an anti PD-L1, as a first-line treatment in cisplatin–ineligible patients with mUC and FGFR mRNA overexpression. The ORR was 44%, with a DCR of 68% and the duration of response was not reached. The most common treatment-emergent events were diarrhea (58%), hyperphosphatemia (45%) and urinary tract infection (36%). The presence of resistance gene mutations was analyzed, and three patients with detectable mutations in PI3K had no objective response [25].
Pemigatinib is another potent and competitive oral inhibitor of the kinase activity of FGFRs 1, 2 and 3. There was a phase II clinical trial (FIGHT-201) with mUC patients who progressed on one or several lines of therapy or were platinum ineligible [26]. Sixty-four patients with some FGFR3 mutation or fusion were assigned to cohort A, and 36 patients with other FGF/FGFR genetic mutations were assigned to cohort B and received pemigatinib. ORR was 25% (95% CI, 14–40%). The efficacy of pemigatinib in combination with pembrolizumab was compared with the standard of care (CT or IT) in patients with cisplatin-ineligible UC in a phase II randomized study (FIGHT-205, NCT04003610).
TAS-120 is a selective irreversible inhibitor for FGFR 1–4. A phase I study treated 134 patients with different advanced solid tumors and FGFR aberrations. Twenty-one mUC patients were included. In the dose-escalation phase, a 20 mg per day oral dose of TAS-120 was considered safe and exhibited clinical activity in various tumors, which need to be confirmed [27].
Debio-1347 is a small oral molecule that selectively inhibits the ATP binding site of FGFR1–3. A phase I clinical trial evaluated the safety and antitumor activity of debio-1347 in 58 patients with solid tumors with FGFR 1–3 alterations; 10% of patients had mUC [28].
Dovitinib is a small multikinase inhibitor that binds to FGFR3, inhibiting its phosphorylation. A phase II trial was prematurely closed because the ORR was 0% in FGFR3-mutated and FGFR3 wild-type patients [29]. Dovitinib in patients with localized UC did not show a clinical benefit in a phase II trial [30].
Derazantinib is a potent ATP competitive multikinase inhibitor of FGFR 1–3 and the colony stimulating factor 1 receptor (CSF1R) kinase. FIDES-02 is a clinical trial that is evaluating the safety and antitumor activity of single-agent derazantinib or in combination with atezolizumab in patients with mUC and FGFR aberrations (NCT04045613).
Recently, the BISCAY study (NCT02546661), characterized as an ambitious study on prospectively adapting the treatment based on genetic alterations, did not achieve a significant benefit for the patients included. Thus, in patients with FGFR, homologous repair gene or mTOR alterations, the study failed to significantly improve the ORR of 27.6% with durvalumab alone compared to AZD4547+durvalumab (ORR = 28.6%), olaparib+durvalumab (ORR = 35.7%) or vistusertib+durvalumab (ORR = 24.1%) [31].
In general, FGFR inhibitors share some adverse events (AEs) which are most easily manageable, but that require close physical examination monitoring, ophthalmic evaluation and early supportive therapy when required (Table 3) [32].
Table 3. Most common FGFR inhibitor-associated adverse events (AEs).
| Drug | AEs Any Grade (%) | AEs Grade 3/4 (%) |
|---|---|---|
| Erdafitinib | Hyperphosphatemia (77%) | Hyponatremia (11%) Stomatitis (10%) Asthenia (7%) Nail dystrophy (6%) Hand-foot syndrome (5%) |
| Stomatitis (58%) | ||
| Diarrhea (51%) | ||
| Dry mouth (46%) | ||
| Central serous retinopathy (27%) | ||
| Onycholysis (18%) | ||
| Infigratinib | Hyperphosphatemia (46.3%) | Hyperlipasemia (10.4%) Fatigue (7.5%) Anemia (7.5%) Hand-foot syndrome (7.5%) Hypophosphatemia (7.5%) |
| Increase in serum creatinine (41.8%) | ||
| Constipation (37.3%) | ||
| Fatigue (37.3%) | ||
| Anemia (35.8%) | ||
| Rogaratinib | Hyperphosphatemia (60%) | Fatigue (9%) Anemia (6%) Urinary tract infection (8%) Hyperlipasemia (8%) |
| Diarrhea (49%) | ||
| Decreased appetite (36%) | ||
| Fatigue (24%) | ||
| Nausea (28%) | ||
| Urinary tract infection (11%) | ||
| Pemigatinib | Diarrhea (40%) | Urinary tract infection (7%) Fatigue (6%) |
| Alopecia (32%) | ||
| Fatigue (29%) | ||
| Constipation (28%) | ||
| Dry mouth (28%) | ||
| Debio-1347 | Hyperphosphatemia (76%) | Hyperphosphatemia (21%) Anemia (12%) Dyspnea (5%) ALT increased (3%) Stomatitis (3%) |
| Diarrhea (41%) | ||
| Nausea (40%) | ||
| Fatigue (40%) | ||
| Constipation (38%) | ||
| Decreased appetite (33%) | ||
| Nail changes (31%) |
References
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a Promising Druggable Target in Cancer: Molecular Biology and New Drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267, doi:10.1016/j.critrevonc.2017.02.018.
- Farrell, B.; Breeze, A.L. Structure, Activation and Dysregulation of Fibroblast Growth Factor Receptor Kinases: Perspectives for Clinical Targeting. Biochem. Soc. Trans. 2018, 46, 1753–1770, doi:10.1042/BST20180004.
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor Signaling Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266, doi:10.1002/wdev.176.
- Rodriguez-Vida, A.; Saggese, M.; Hughes, S.; Rudman, S.; Chowdhury, S.; Smith, N.R.; Lawrence, P.; Rooney, C.; Dougherty, B.; Landers, D.; et al. Complexity of FGFR Signalling in Metastatic Urothelial Cancer. J. Hematol. Oncol. J. Hematol. Oncol. 2015, 8, 119, doi:10.1186/s13045-015-0221-6.
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast Growth Factor Receptor Signaling in Hereditary and Neoplastic Disease: Biologic and Clinical Implications. Cancer Metastasis Rev. 2015, 34, 479–496, doi:10.1007/s10555-015-9579-8.
- Cihoric, N.; Savic, S.; Schneider, S.; Ackermann, I.; Bichsel-Naef, M.; Schmid, R.A.; Lardinois, D.; Gugger, M.; Bubendorf, L.; Zlobec, I.; et al. Prognostic Role of FGFR1 Amplification in Early-Stage Non-Small Cell Lung Cancer. Br. J. Cancer 2014, 110, 2914–2922, doi:10.1038/bjc.2014.229.
- Katoh, M. Fibroblast Growth Factor Receptors as Treatment Targets in Clinical Oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122, doi:10.1038/s41571-018-0115-y.
- Katoh, M. Therapeutics Targeting FGF Signaling Network in Human Diseases. Trends Pharmacol. Sci. 2016, 37, 1081–1096, doi:10.1016/j.tips.2016.10.003.
- Di Martino, E.; Tomlinson, D.C.; Williams, S.V.; Knowles, M.A. A Place for Precision Medicine in Bladder Cancer: Targeting the FGFRs. Future Oncol. 2016, 12, 2243–2263, doi:10.2217/fon-2016-0042.
- Wang, L.; Šuštić, T.; Leite de Oliveira, R.; Lieftink, C.; Halonen, P.; van de Ven, M.; Beijersbergen, R.L.; van den Heuvel, M.M.; Bernards, R.; van der Heijden, M.S. A Functional Genetic Screen Identifies the Phosphoinositide 3-Kinase Pathway as a De-terminant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur. Urol. 2017, 71, 858–862, doi:10.1016/j.eururo.2017.01.021.
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour Cell Responses to New Fibroblast Growth Factor Receptor Tyrosine Kinase Inhibitors and Identification of a Gatekeeper Mutation in FGFR3 as a Mechanism of Acquired Resistance. Oncogene 2013, 32, 3059–3070, doi:10.1038/onc.2012.319.
- Research, C. for D.E. and FDA Grants Accelerated Approval to Erdafitinib for Metastatic Urothelial Carcinoma. FDA 2019.
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348, doi:10.1056/NEJMoa1817323.
- Siefker-Radtke, A.O.; Necchi, A.; Park, S.H.; García-Donas, J.; Huddart, R.A.; Burgess, E.F.; Fleming, M.T.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. ERDAFITINIB in Locally Advanced or Metastatic Urothelial Carcinoma (MUC): Long-Term Outcomes in BLC2001. J. Clin. Oncol. 2020, 38, 5015, doi:10.1200/JCO.2020.38.15_suppl.5015.
- Janssen Research & Development, LLC. A Phase 3 Study of Erdafitinib Compared With Vinflunine or Docetaxel or Pem-brolizumab in Subjects With Advanced Urothelial Cancer and Selected FGFR Gene Aberrations. Available online: clinicaltri-als.gov (accessed on 2020).
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25, doi:10.1016/j.cell.2017.09.007.
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non–T-Cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568, doi:10.1158/2326-6066.CIR-15-0274.
- Moreno, V.; Loriot, Y.; Rutkowski, P.; Beato, C.; Felip, E.; Duran, I.; Kowalski, D.; Siena, S.; Cortinovis, D.; Geoffrois, L.; et al. Evolving Development of PD-1 Therapy: Cetrelimab (JNJ-63723283) from Monotherapy to Combination with Erdafitinib. J. Clin. Oncol. 2020, 38, 3055–3055, doi:10.1200/JCO.2020.38.15_suppl.3055.
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.M.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165, doi:10.1200/JCO.2016.67.2048.
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821, doi:10.1158/2159-8290.CD-18-0229.
- Dizman, N.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Quinn, D.I.; Petrylak, D.P.; Galsky, M.D.; Vaishampayan, U.N.; De Giorgi, U.; Gupta, S.; Burris, H.A.; et al. Infigratinib in Upper Tract Urothelial Carcinoma vs Urothelial Carcinoma of the Bladder and Association with Comprehensive Genomic Profiling/Cell-Free DNA Results. J. Clin. Oncol. 2019, 37, 4510 doi:10.1200/JCO.2019.37.15_suppl.4510.
- Pal, S.K.; Bajorin, D.; Dizman, N.; Hoffman-Censits, J.; Quinn, D.I.; Petrylak, D.P.; Galsky, M.D.; Vaishampayan, U.; De Giorgi, U.; Gupta, S.; et al. Infigratinib in Upper Tract Urothelial Carcinoma versus Urothelial Carcinoma of the Bladder and Its As-sociation with Comprehensive Genomic Profiling and/or Cell-Free DNA Results. Cancer 2020, 126, 2597–2606, doi:10.1002/cncr.32806.
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; Soo, R.A.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in Patients with Advanced Cancers Selected by FGFR MRNA Expression: A Phase 1 Dose-Escalation and Dose-Expansion Study. Lancet Oncol. 2019, 20, 1454–1466, doi:10.1016/S1470-2045(19)30412-7.
- Quinn, D.I.; Petrylak, D.P.; Bellmunt, J.; Necchi, A.; Gurney, H.; Lee, J.-L.; Van Der Heijden, M.S.; Rosenbaum, E.; Penel, N.; Pang, S.-T.; et al. FORT-1: Phase II/III Study of Rogaratinib versus Chemotherapy (CT) in Patients (Pts) with Locally Advanced or Metastatic Urothelial Carcinoma (UC) Selected Based on FGFR1/3 MRNA Expression. J. Clin. Oncol. 2020, 38, 489–489, doi:10.1200/JCO.2020.38.6_suppl.489.
- Rosenberg, J.E.; Gajate, P.; Morales-Barrera, R.; Lee, J.-L.; Necchi, A.; Penel, N.; Zagonel, V.; Sierecki, M.R.; Piciu, A.-M.; Ellinghaus, P.; et al. Safety and Preliminary Efficacy of Rogaratinib in Combination with Atezolizumab in a Phase Ib/II Study (FORT-2) of First-Line Treatment in Cisplatin-Ineligible Patients (Pts) with Locally Advanced or Metastatic Urothelial Cancer (UC) and FGFR MRNA Overexpression. J. Clin. Oncol. 2020, 38, 5014–5014, doi:10.1200/JCO.2020.38.15_suppl.5014.
- Necchi, A.P.D. Interim Results of Fight-201, a Phase 2, Open-Label, Multicenter Study of INCB054828 in Patients (Pts) with Metastatic or Surgically Unresectable Urothelial Carcinoma (UC) Harboring Fibroblast Growth Factor (FGF)/FGF Receptor (FGFR) Genetic Alterations (GA). Ann. Oncol. 2018, 29, Viii303-Viii331
- Meric-Bernstam, F.; Goyal, L.; Tran, B.; Matos, I.; Arkenau, H.-T.; He, H.; Huang, J.; Bahleda, R. Abstract CT238: TAS-120 in Patients with Advanced Solid Tumors Bearing FGF/FGFR Aberrations: A Phase I Study. In Proceedings of the Clinical Trials, AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019; p. CT238.
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-Escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Pa-tients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res. 2019, 25, 2699–2707, doi:10.1158/1078-0432.CCR-18-1959.
- Milowsky, M.I.; Dittrich, C.; Durán, I.; Jagdev, S.; Millard, F.E.; Sweeney, C.J.; Bajorin, D.; Cerbone, L.; Quinn, D.I.; Stadler, W.M.; et al. Phase 2 Trial of Dovitinib in Patients with Progressive FGFR3-Mutated or FGFR3 Wild-Type Advanced Urothelial Carcinoma. Eur. J. Cancer 1990 2014, 50, 3145–3152, doi:10.1016/j.ejca.2014.10.013.
- Hahn, N.M.; Bivalacqua, T.J.; Ross, A.E.; Netto, G.J.; Baras, A.; Park, J.C.; Chapman, C.; Masterson, T.A.; Koch, M.O.; Bihrle, R.; et al. A Phase II Trial of Dovitinib in BCG-Unresponsive Urothelial Carcinoma with FGFR3 Mutations or Overexpression: Hoosier Cancer Research Network Trial HCRN 12-157. Clin. Cancer Res. 2017, 23, 3003–3011, doi:10.1158/1078-0432.CCR-16-2267.
- Powles, T.; Balar, A.; Gravis, G.; Jones, R.; Ravaud, A.; Florence, J.; Grivas, P.; Petrylak, D.P.; Galsky, M.; Carles, J.; et al. An Adaptive, Biomarker Directed Platform Study in Metastatic Urothelial Cancer (BISCAY) with Durvalumab in Combination with Targeted Therapies. Ann. Oncol. 2019, 30, v356–v357, doi:10.1093/annonc/mdz249.001.
- Mahipal, A.; Tella, S.H.; Kommalapati, A.; Yu, J.; Kim, R. Prevention and Treatment of FGFR Inhibitor-Associated Toxicities. Crit. Rev. Oncol. Hematol. 2020, 155, 103091, doi:10.1016/j.critrevonc.2020.103091.



