 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kenneth K. Wu | + 2573 word(s) | 2573 | 2021-01-21 10:04:29 | | | |
| 2 | Vicky Zhou | -1513 word(s) | 1060 | 2021-01-26 03:27:15 | | |
Video Upload Options
Cellular senescence is a hallmark of aging. Accumulation of senescent cells promotes aging and triggers age-related disorders.
1. Introduction
Cellular senescence was originally observed in cultured fibroblasts following limited replications [1]. It was subsequently noted as a response to DNA damage, telomere attrition, mitochondrial dysfunction, and oncogenic, hyperglycemic, and oxidative stresses [2][3][4]. Cellular senescence plays an important role in parturition and embryo development [5][6]. It may influence the fate of tumorigenesis through senescence-associated secretory phenotype (SASP). It was reported that acute senescent cells induce immortalized prostate cells to undergo senescence via SASP but have no effect on metastatic prostate cancer cells [7]. Replicative, developmental, and stress-induced premature cell senescence share common cellular phenotypic changes including an increased expression of p16 and p21, cell cycle and proliferation arrest, senescence-associated (SA) heterochromatin foci, SA-β galactosidase (β gal), and SASP, as well as cellular morphological changes [8]. Phenotypic changes of senescent cells are mediated by multiple signaling pathways leading to complex transcriptional reprogramming [9][10].
Hyperglycemia due to type 2 diabetes (T2D) and pre-diabetic metabolic syndrome and obesity has emerged as a key extracellular stress signal to induce cellular senescence as well as cell death [11]. As T2D is increasing with aging and contributes to age-related chronic diseases [11], hyperglycemia has become a leading age stress factor. Hyperglycemia induces cellular senescence through metabolism shift, reactive oxygen species (ROS) generation, mitochondrial dysfunction, and aberrant gene expressions.
Replicative and stress (hyperglycemia and oxidative stress)-induced mesenchymal stromal cell senescence has been extensively investigated as it is critical for MSC-based cell therapy. Mesenchymal stromal cells (MSCs) are isolated and characterized according to a set of criteria [12][13]. Current isolation procedures generate heterogeneous nonclonal stromal cell populations with different multipotent and differentiation potentials [14]. MSCs possess immunosuppressive and anti-inflammatory properties [15][16]. As MSCs can be obtained and cultured with ease, they are popular sources for cell-based therapy of a variety of human diseases. More than 700 clinical trials have been registered [14]. However, MSC-based cell therapy faces challenging problems. Replicative senescence of cultured MSCs limits the cell expansion and its availability for cell therapy. Moreover, stress-induced premature senescence in vitro and in vivo reduces the efficacy of transplanted MSCs in tissue regeneration and treatment of autoimmune and inflammatory diseases. New strategies are actively being employed to develop new drugs to combat cellular senescence. The candidate drugs are either senolytic, which kill and remove senescent cells, or senomorphic, which modify senescent cell phenotypes to attenuate their tissue-damaging effects [17][18][19]. Senomorphic agents comprise a wide range of compounds with different targets aiming at reducing SASP and senescent markers without causing cell apoptosis [18]. Recent studies indicate that tryptophan metabolites produced via the tryptophan hydroxylase (TPH) pathway defend against replicative and hyperglycemia or oxidative stress-induced cell senescence. 5-methoxytryptophan (5-MTP) was reported to rescue bone marrow mesenchymal stromal cells (BM-MSCs) from high glucose (HG)-induced senescence [20], while melatonin protects MSC from replicative and stress-induced senescence [21]. Melatonin and 5-MTP represent a new class of senomorphic compounds which may be useful in protecting MSC against senescence and age-related diseases.
2. Hyperglycemia Induces Cellular Senescence
High blood glucose levels (hyperglycemia) contribute to diabetic microvascular, renal, retinal and neural complications by multiple mechanisms including mitochondrial dysfunction and ROS generation [22][23]. Results from in vivo and in vitro experiments have shown that hyperglycemia induces cellular damage, apoptosis, and necrosis through ROS generation and mitochondrial dysfunction [22][23]. In addition, hyperglycemia was reported to induce renal tubular cell and retinal endothelial cell senescence in streptozotocin-induced diabetic mice [24][25] and HG in cultured media was reported to induce senescence of diverse cell types including MSCs [26][27][28]. Senescent cells cause further tissue damage through secretion of pro-inflammatory cytokines and proteolytic enzymes [29][30].
2.1. HG-Induced Cellular Senescence Is Attributed to Mitochondrial Dysfunction and ROS Generation
The exact mechanisms by which HG induces senescence are not entirely clear. Mitochondrial dysfunction and ROS generation are considered to be major players. HG induces mitochondrial ROS generation by enhancing mitochondrial metabolism via tricarboxylic acid (TCA) cycle and oxidative phosphorylation [22]. ROS generation is closely related to mitochondrial morphological changes. It was reported that HG-treated rat liver cells undergo mitochondrial fission, which was required for ROS generation [31]. ROS, in turn, cause mitochondrial fission [32], creating a vicious cycle (Figure 1). It was also reported that HG increases ROS through activation of NADPH oxidase [33][34], but its relevance to cell senescence is unclear and remains to be investigated. ROS overproduction is considered to be a major cause of cell damage and lethality. However, at sublethal concentrations, H2O2 induces cellular senescence as a way of protecting cells from ROS-induced death [35][36]. ROS represent a common mediator via which diverse stress signals induce cellular senescence. For example, Ras overexpression in fibroblasts induce cellular senescence by elevation of ROS generation [36].
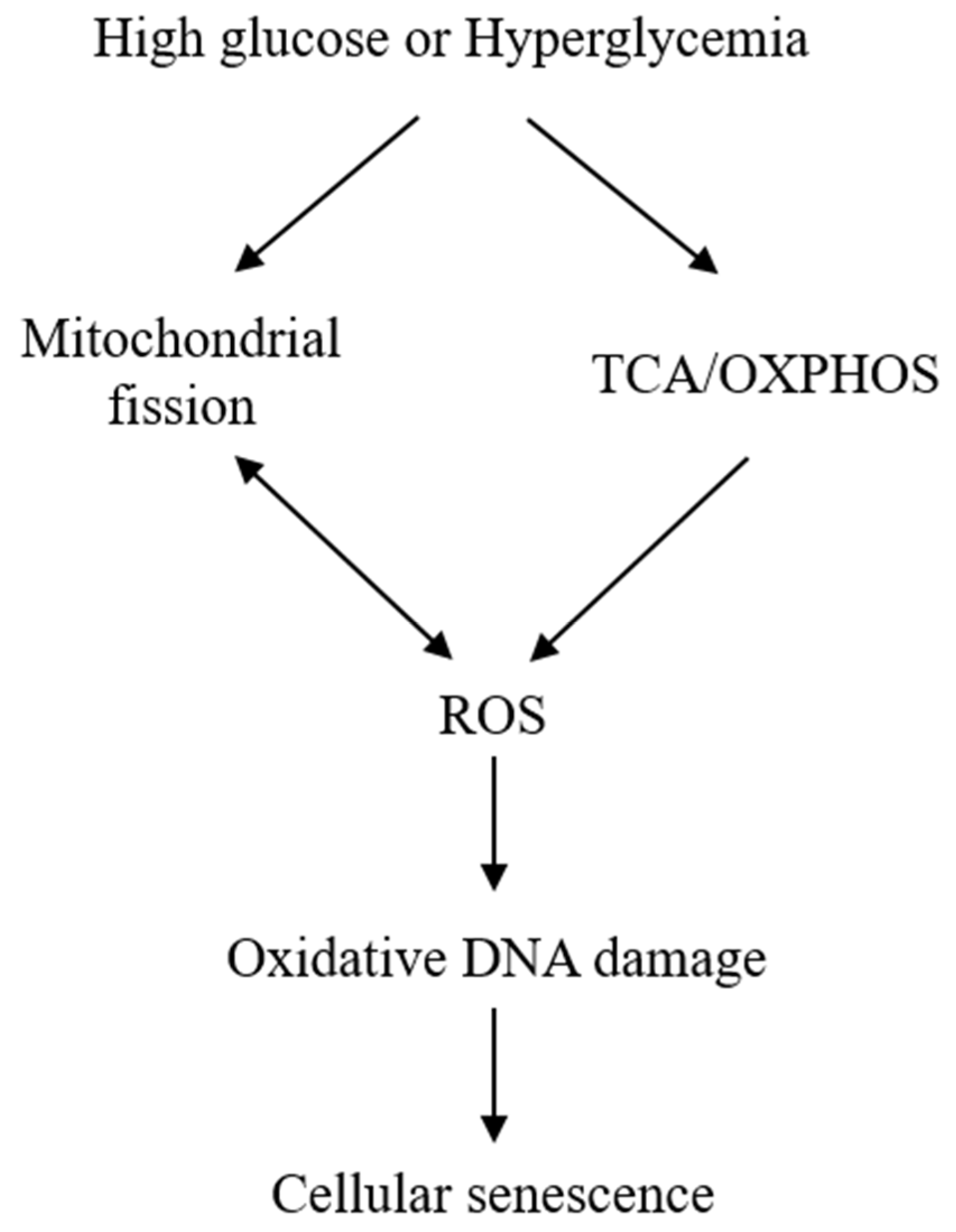
ROS induces cellular senescence by oxidative damage to DNA, leading to telomere attrition and altered expression of p53, p16, and p21 [37][38]. However, increased ROS generation cannot explain all the phenotypic manifestations. Mitochondrial structural changes and functional defects contribute significantly to stress-induced senescence [39]. Changes in mitochondrial dynamics [40][41], metabolism, and signaling molecules such as AMPK [42][43][44] are considered to mediate senescence independent of ROS.
2.2. HG Induces MSC Senescence
MSCs in bone marrow reside in hypoxic microenvironment. They depend on glycolysis as energy source and thus express relatively low levels of oxidative phosphorylation (OXPHOS) proteins [45]. MSCs cultured in nutrient-rich normoxic conditions shifts the metabolism to OXPHOS [46]. MSC metabolism is altered during osteoblastic vs. chondrogenic differentiation. Osteoblastic differentiation requires OXPHOS, while chondrogenic differentiation uses aerobic glycolysis in energy generation [47]. When MSCs are incubated with HG medium, excessive intracellular glucose shifts metabolism from aerobic glycolysis to TCA cycle and OXPHOS. Consequently, a high level of ROS is leaked from the electron transport chain which induces MSC senescence [48][49]. Effects of HG on MSC mitochondrial biogenesis and metabolism have not been described. However, it is likely HG-induced mitochondrial structural and metabolic changes contribute to MSC senescence. MSC senescence puts a limit to MSC expansion, which hampers its use in cell therapy. Furthermore, it impairs MSC function which reduces its support for hematopoiesis and immunosuppressive properties .
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Cell Res. 1961, 25, 585–621, doi:10.1016/0014-4827(61)90192-6.
- Rodier, F.; Campisi, J. Four faces of cellular senescence. Cell Biol. 2011, 192, 547–556, doi:10.1083/jcb.201009094.
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Med. 2015, 21, 1424–1435, doi:10.1038/nm.4000.
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114, doi:10.1101/gad.235184.113.
- Behnia, F.; Taylor, B.D.; Woodson, M.; Kacerovsky, M.; Hawkins, H.; Fortunato, S.J.; Saade, G.R.; Menon, R. Chorioamniotic membrane senescence: A signal for parturition? J. Obstet. Gynecol. 2015, 213, 359-e1, doi:10.1016/j.ajog.2015.05.041.
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130, doi:10.1016/j.cell.2013.10.041.
- Alessio, N.; Aprile, D.; Squillaro, T.; Di Bernardo, G.; Finicelli, M.; Melone, M.A.; Peluso, G.; Galderisi, U. The Senescence-Associated Secretory Phenotype (SASP) from mesenchymal stromal cells impairs growth of immortalized prostate cells but has no effect on metastatic prostatic cancer cells. Aging 2019, 11, 5817–5828, doi:10.18632/aging.102172.
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Rev. Mol. Cell Biol. 2007, 8, 729–740, doi:10.1038/nrm2233.
- Alfego, D.; Rodeck, U.; Kriete, A. Global mapping of transcription factor motifs in human aging. PLoS ONE 2018, 13, e0190457, doi:10.1371/journal.pone.0190457.
- Lanigan, F.; Geraghty, J.G.; Bracken, A.P. Transcriptional regulation of cellular senescence. Oncogene 2011, 30, 2901–2911, doi:10.1038/onc.2011.34.
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298, doi:10.2337/db14-1820.
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A.; International Society for Cellular Therapy. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395, doi:10.1080/14653240500319234.
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317, doi:10.1080/14653240600855905.
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transpl. 2016, 25, 829–848, doi:10.3727/096368915X689622.
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal Stem Cells in health and disease. Rev. Immunol. 2008, 8, 726–736, doi:10.1038/nri2395.
- English, K. Mechanisms of mesenchymal stromal cell immunomodulation. Cell Biol. 2013, 91, 19–26, doi:10.1038/icb.2012.56.
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Med. 2018, 24, 1246–1256, doi:10.1038/s41591-018-0092-9.
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Rev. Drug Discov. 2017, 16, 718–735, doi:10.1038/nrd.2017.116.
- Liu, J.; Ding, Y.; Liu, Z.; Liang, X. Senescence in Mesenchymal Stem Cells: Functional Alterations, Molecular Mechanisms, and Rejuvenation Strategies. Cell Dev. Biol. 2020, 8, 258, doi:10.3389/fcell.2020.00258.
- Chang, T.C.; Hsu, M.F.; Shih, C.Y.; Wu, K.K. 5-methoxytryptophan protects MSCs from stress induced premature senescence by upregulating FoxO3a and mTOR. Rep. 2017, 7, 11133, doi:10.1038/s41598-017-11077-4.
- Lee, J.H.; Yoon, Y.M.; Song, K.H.; Noh, H.; Lee, S.H. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L-mitophagy pathway. Aging Cell 2020, 19, e13111, doi:10.1111/acel.13111.
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820, doi:10.1038/414813a.
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625, doi:10.2337/diabetes.54.6.1615.
- Kitada, K.; Nakano, D.; Ohsaki, H.; Hitomi, H.; Minamino, T.; Yatabe, J.; Felder, R.A.; Mori, H.; Masaki, T.; Kobori, H.; et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. Diabetes Complicat. 2014, 28, 604–611, doi:10.1016/j.jdiacomp.2014.05.010.
- Shosha, E.; Xu, Z.; Narayanan, S.P.; Lemtalsi, T.; Fouda, A.Y.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. J. Mol. Sci. 2018, 19, 1215, doi:10.3390/ijms19041215.
- Blazer, S.; Khankin, E.; Segev, Y.; Ofir, R.; Yalon-Hacohen, M.; Kra-Oz, Z.; Gottfried, Y.; Larisch, S.; Skorecki, K.L. High glucose-induced replicative senescence: Point of no return and effect of telomerase. Biophys. Res. Commun. 2002, 296, 93–101, doi:10.1016/s0006-291x(02)00818-5.
- Zhong, W.; Zou, G.; Gu, J.; Zhang, J. L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res. Clin. Pract. 2010, 89, 38–45, doi:10.1016/j.diabres.2010.03.013.
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: Role of superoxide production by NADPH oxidase. PLoS ONE 2015, 10, e0123169, doi:10.1371/journal.pone.0123169.
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. Cell Biol. 2018, 217, 65–77, doi:10.1083/jcb.201708092.
- Palmer, A.K.; Gustafson, B.; Kirkland, J.L.; Smith, U. Cellular senescence: At the nexus between ageing and diabetes. Diabetologia 2019, 62, 1835–1841, doi:10.1007/s00125-019-4934-x.
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Natl. Acad. Sci. USA 2006, 103, 2653–2658, doi:10.1073/pnas.0511154103.
- Abuarab, N.; Munsey, T.S.; Jiang, L.H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn 2+-mediated mitochondrial fission. Signal. 2017, 10, eaal4161, doi:10.1126/scisignal.aal4161.
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through Protein Kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945, doi:10.2337/diabetes.49.11.1939.
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Des Rosiers, C.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Res. 2011, 9, 237–246, doi:10.1093/cvr/cvr230.
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: P53 and Rb control G1 arrest but not cell replication. J. 1998, 332, 43–50, doi:10.1042/bj3320043.
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of Reactive Oxygen Species. Biol. Chem. 1999, 274, 7936–7940, doi:10.1074/jbc.274.12.7936.
- S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in P53-mediated cell fate decisions. Mol. Cell Biol. 2003, 23, 8576–8585, doi:10.1128/mcb.23.23.8576-8585.2003.
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Natl. Acad. Sci. USA 1995, 92, 4337–4341, doi:10.1073/pnas.92.10.4337.
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7, doi:10.1111/acel.12287.
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. Biol. Chem. 2005, 280, 26185–26192, doi:10.1074/jbc.M503062200.
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant Mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. Cell Physiol. 2006, 209, 468–480, doi:10.1002/jcp.20753.
- Stöckl, P.; Hütter, E.; Zwerschke, W.; Jansen-Dürr, P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human Fibroblasts. Gerontol. 2006, 41, 674–682, doi:10.1016/j.exger.2006.04.009.
- Borradaile, N.M.; Pickering, J.G. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009, 8, 100–112, doi:10.1111/j.1474-9726.2009.00453.x.
- Ho, C.; van der Veer, E.; Akawi, O.; Pickering, J.G. SIRT1 markedly extends replicative lifespan if the NAD+ salvage pathway is enhanced. FEBS Lett. 2009, 583, 3081–3085, doi:10.1016/j.febslet.2009.08.031.
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968, doi:10.1634/stemcells.2007-0509.
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Rev. Mol. Cell Biol. 2014, 15, 243–256, doi:10.1038/nrm3772.
- Pattappa, G.; Heywood, H.K.; de Bruijn, J.D.; Lee, D.A. The metabolism of human Mesenchymal Stem Cells during proliferation and differentiation. Cell Physiol. 2011, 226, 2562–2570, doi:10.1002/jcp.22605.
- Stolzing, A.; Coleman, N.; Scutt, A. Glucose-induced replicative senescence in Mesenchymal Stem Cells. Rejuvenation Res. 2006, 9, 31–35, doi:10.1089/rej.2006.9.31.
- Zhang, D.; Lu, H.; Chen, Z.; Wang, Y.; Lin, J.; Xu, S.; Zhang, C.; Wang, B.; Yuan, Z.; Feng, X.; et al. High glucose induces the aging of Mesenchymal Stem Cells via Akt/mTOR signaling. Med. Rep. 2017, 16, 1685–1690, doi:10.3892/mmr.2017.6832.

