 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Abhishek Sinha | + 1751 word(s) | 1751 | 2021-01-20 08:42:17 | | | |
| 2 | Camila Xu | Meta information modification | 1751 | 2021-01-25 10:22:32 | | |
Video Upload Options
Transforming growth factor β (TGFβ) is a secreted growth and differentiation factor that influences vital cellular processes like proliferation, adhesion, motility, and apoptosis.
1. Introduction
Transforming growth factor-beta (TGFβ) was discovered more than thirty years ago as a secreted polypeptide from sarcoma virus-infected cells that promoted the soft agar independent growth of normal rat kidney (NRK) cells [1][2]. This biological property was associated with oncogenes, hence the name transforming growth factor. However, studies shortly thereafter discovered that this effect of TGFβ (unlike the effect of oncogenes) was reversible, and that TGFβ can have potent cell growth inhibitory effects [3]. Thirty-three human genes encoding distinct but closely structurally and functionally related TGFβ family members have been identified, which includes TGFβ1,-β2 and -β3, activins, inhibins, bone morphogenetic proteins (BMPs), myostatin, nodal and the mullerian inhibitory substance (MIS) [4][5][6]. The active TGFβ molecule is a dimer stabilized by hydrophobic interactions and a disulfide bond [7]. Following the binding of TGFβ to its specific receptors that are expressed on nearly all cell types, TGFβ regulates a plethora of biological processes, ranging from cell proliferation and differentiation, embryogenesis, hormonal synthesis and secretion, immunity to tissue remodeling and repair [6][8]. Because of its ability to induce cell growth inhibition and apoptosis in normal and pre-malignant cells, TGFβ has been described as a potent tumor suppressor [8][9]. In support of this notion, mutations in the components of the TGFβ signaling cascade have been identified in a number of human cancers, including hereditary nonpolyposis colon cancer, hepatocellular carcinoma (HCC), and pancreatic and ovarian cancer [10]. In contrast to this activity, TGFβ can also function as a tumor promoter by promoting cancer cell proliferation, stimulating epithelial-to-mesenchymal transition (EMT) and migration of cancer cells, and indirectly by acting on the tumor microenvironment, promoting angiogenesis, and/or immune evasion in advanced stages of tumor progression [8][9]. Active TGFβ induces the heteromeric complex formation of two single transmembrane serine/threonine kinase family receptors, i.e., TGFβ type I and type II receptor (TβRI and TβRII, respectively) [11][12]. Two molecules of TβRII associate with two TβRIs molecules thereby forming a tetrameric receptor complex [13][14]. Upon ligand-induced complex formation, the TβRII’s constitutively active kinase phosphorylates TβRI [also termed activin receptor-like kinase-5 (ALK5)] on serine and threonine residues in the juxta membrane glycine-serine residue-rich (GS) domain [15][16]. TβRI acts downstream of TβRII, and is the main component of the receptor complex that triggers various downstream signaling activities [17]. The signal is relayed from the receptors to the nucleus via cytoplasmic effector molecules, a process called SMAD and non-SMAD signaling [18][19].The action of TGFβ is highly context dependent and is subject to positive and negative regulation in each step of the signaling pathway [6][20]. This is needed to enable spatio-temporal control and allow for crosstalk with other signaling pathways [21]. An important mechanism by which the expression, localization, and activity of TGFβ signaling components is controlled is by post-translational modification (PTMs). For example, PTMs that occur in receptors and SMAD proteins include phosphorylation, ubiquitination, sumoylation, ribosylation, and acetylation [22][23][24][25][26][27][28][29]. In particular, ubiquitination is emerging as a key regulatory mechanism by which intracellular signaling intensity, duration, and specificity is reversibly controlled through the action of multiple E3 ubiquitin ligases and deubiquitinating enzymes [30][31][32]. A concept is emerging that disruption of the ubiquitin modification and function of TGFβ pathway components may result in illicit protein activity leading towards human diseases including cancer.
2. TGFβ as Tumor Suppressor and Tumor Promoter
In normal (healthy) and premalignant cells, the TGFβ signaling pathway prompts a tumor suppressive role. However, in advanced metastatic tumors, this pathway can be blunted or corrupted or even utilized by cancer cells to promote oncogenic functions [9][61] (Figure 2).
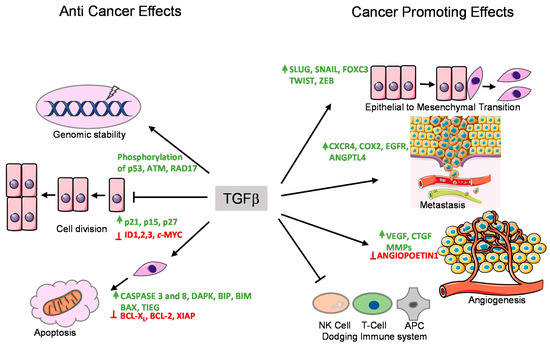
Figure 2. Tumor suppressor and promoting effects of the TGFβ pathway. TGFβ exerts its anti-tumor and tumor promoting effects by regulating the expression of various target genes. These functions are performed by various means shown with arrows. The genes whose expression are enhanced by TGFβ are shown in green, whereas the genes whose expression are suppressed by TGFβ are marked in red.
TGFβ induces cell cycle arrest in G1 phase by inducing the expression of cell cycle inhibitory proteins, like cyclin-dependent kinase inhibitors (CDKIs) p15INK4B and/or p21KIP1, which in turn inhibit specific CDK functions [62][63]. TGFβ also represses the expression of growth inducing factors such as the oncogene c-MYC, and the ID family of transcription factors (ID1, ID2, and ID3), which also results in inhibition of cell proliferation [64][65][66][67]. Another anti-tumor function of TGFβ is its role in promoting apoptosis (Figure 2). In hepatocytes and B-lymphocytes, TGFβ promotes SMAD and the p38 MAPK-dependent transcriptional induction of the pro-apoptotic Bcl-2 family members BMF and BIM. These in turn induce the pro-apoptotic factor BAX, which leads to mitochondrial release of Cytochrome C and increase in caspase-dependent apoptosis [68][69]. Similarly, TGFβ signaling is known to repress BCL-XL and anti-apoptotic BCL-2 family members leading to promotion of apoptosis [70][71][72]. In liver, TGFβ promotes the expression of pro-apoptotic protein, death-associated protein kinase (DAPK), in a SMAD dependent manner [73] (Figure 2).
In mice models, TGFβ has been found to regulate genomic stability [74][75][76] (Figure 2). Knockout of TβR1inhibited the phosphorylation of ataxia-telangiectasia mutated (ATM), p53, CHK2, and RAD17, which increased the radiation-sensitivity of cells derived from knockout mice compared to normal mice [77]. Similarly, SMAD4 conditional knockout mice with head and neck cancer, revealed a role for SMAD4 in the regulation of the Fanconi anemia/BRCA DNA repair pathway, also suggesting an involvement in genomic stability [78]. The same study demonstrated a correlation of SMAD4 knockdown with the downregulation of BRCA1 and RAD51 protein expression in head and neck squamous cell carcinoma (HNSCC) as well as increased expression of TβR1 and phospho-SMAD3 [78]. The exact mechanism of this regulation remains to be determined.
Mutation or functional inactivation of TGFβ receptors or downstream SMAD proteins have been associated with progression of malignancies [79], as it mitigates the TGFβ/SMAD-induced cytostatic responses, and leads to cell hyperproliferation. In particular, TβRI, TβRII, SMAD2, and SMAD4 are frequently mutated, deleted, or attenuated (gene/loss of heterozygosity/expression) in certain cancer subtypes. Inactivating mutations or deletions of genes encoding TGFβ receptors and SMADs are common in esophageal, colorectal, and pancreatic adenocarcinomas. SMAD4 inactivating mutations are common in gastric, colorectal, and pancreatic adenocarcinomas, whereas up to 20% of head and neck, bladder, cervical, and lung squamous carcinomas contain inactivating mutations in TGFβ signaling components like SMAD2, SMAD3, SMAD4, TβR1, or TβRII [8].
In late stages of cancer, TGFβ signaling can switch towards a tumor promoting function through diverse mechanisms (Figure 2). In the following section, we discuss the pro-oncogenic role of TGFβ signaling with a few examples. The following roles are specifically true for cases where SMADs or receptors are not functionally inactivated by mutations. The pro-oncogenic responses of TGFβ can be broadly classified into three major groups.
2.1. Epithelial-to-Mesenchymal Transition (EMT) and Invasion
EMT is an important mechanism by which epithelial cells acquire fibroblast-like properties. This is a characteristic feature in normal cells undergoing embryogenesis and wound healing, but also occurs in pathological processes like fibrogenesis and tumorigenesis. TGFβ is a well-known inducer of EMT (Figure 3). Notably, cells that overexpress SMAD7 or have reduced expression of SMAD3/4 show significantly decreased EMT in response to TGFβ [80][81]. TGFβ was found to induce the expression of transcription factors such as SNAIL, SLUG, TWIST, and FOXC3, which are of key importance to downregulate epithelial genes such as E-CDH1 (encoding E-Cadherin) and mediating acquisition of mesenchymal features by cancer cells [80][82]. Acquiring mesenchymal characteristics is a prerequisite for the spread of cancer cells at secondary sites of the body and the process of metastasis. During breast cancer progression, TGFβ induces single cell migration through a SMAD-mediated pathway involving downstream activation of epidermal growth factor receptor (EGFR), Jun and Rho signaling pathways, and connective tissue growth factor (CTGF) [83][84]. TGFβ also stimulates the secretion of the parathyroid hormone related protein (PTHrP), which in turn induces the expression of bone homing receptor C-X-C chemokine receptor type 4 (CXCR4). CXCR4 promotes chemoattraction of the breast cancer cells to the bone secondary sites during metastasis [85][86] (Figure 2).
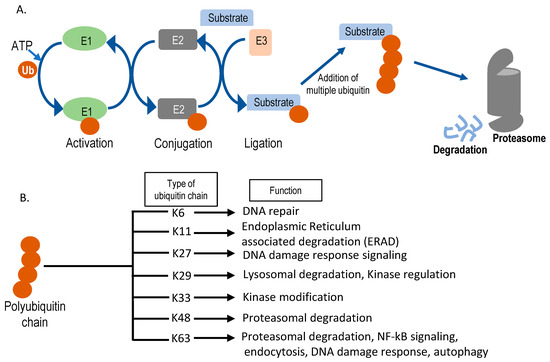
Figure 3. Schematic representation of the ubiquitin-proteasomal degradation pathway. (A) Sequential action of E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin ligase) enzymes in substrate ubiquitination. Ubiquitin is denoted as Ub. After multiple ubiquitin molecules are attached to the substrate forming lysine (K)-48, it can get degraded by the 26S proteasome. (B) Different kinds of polyubiquitin chains formed inside the cell and their cellular functions. Seven different ubiquitin lysine residues can be used for sequential chain formation and, based on the position of chain formation, functions vary.
2.2. Promoting Angiogenesis
Angiogenesis is the process in which new vasculature is formed from existing blood vessels. This is central to the development of tumors as blood vessels provide a steady supply of blood and associated metabolites, cytokines, and growth factors, which promotes uncontrolled cell proliferation. TGFβ can promote angiogenesis through its effects on various angiogenic factors such as vascular endothelial growth factor (VEGF) and CTGF [85][87] (Figure 2). TGFβ also promotes the production and secretion of well-known angiogenesis promoting matrix metalloproteases, MMP-2 and MMP-9 [76][88]. It has been also shown that TβRI-mediated SMAD2/3 signaling promotes transcription of extracellular matrix proteins such as fibronectin and plasminogen activator inhibitor type 1 (PAI1), which regulate angiogenesis by inducing vessel maturation [89]. Thus, TGFβ can promote angiogenesis and cancer progression by promoting expression of a variety of angiogenic factors (Figure 2).
2.3. Immunomodulatory Effects
TGFβ has a broad immunomodulatory effect resulting in mostly pro-tumorigenic effects [90]. In short, the adaptive immune system, consisting of T and B cells, can eliminate tumor cells upon recognition. Through its immunomodulating role, TGFβ signaling can inhibit the function of antigen presenting cells, thereby decreasing T cell activation and decreasing elimination of tumor cells [91]. In addition, TGFβ directly inhibits CD4+ and CD8+ T cells, as well as natural killer (NK) cells. The effect of TGFβ on the innate immune system, like neutrophils and macrophages, also mostly results in pro-tumorigenic effects as it drives these cells from a type 1 differentiated cell into a most immature type 2 cell. This modulation occurs in neutrophils, macrophages, and in the T cells leading to enhanced release of TGFβ into the tumor microenvironment [74][92] (Figure 2).
Here we have discussed, with examples, the pro-tumorigenic effects of TGFβ though it is noteworthy that there are other ways (e.g., regulating stemness, relapse of cancer, etc.) through which this signaling can promote cancer. Overall, it can be concluded that tightly controlled TGFβ signaling is critical for normal functioning of the cell and that perturbations of this pathway can contribute to cancer progression.
References
- Twardzik, D.R.; Brown, J.P.; Ranchalis, J.E.; Todaro, G.J.; Moss, B. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc. Natl. Acad. Sci. USA 1985, 82, 5300–5304, doi:10.1073/pnas.82.16.5300.
- Roberts, A.B.; Anzano, M.A.; Meyers, C.A.; Wideman, J.; Blacher, R.; Pan, Y.C.; Stein, S.; Lehrman, S.R.; Smith, J.M.; Lamb, L.C.; et al. Purification and properties of a type beta transforming growth factor from bovine kidney. Biochemistry 1983, 22, 5692–5698, doi:10.1021/bi00294a002.
- Inagaki, M.; Moustakas, A.; Lin, H.Y.; Lodish, H.F.; Carr, B.I. Growth inhibition by transforming growth factor beta (TGF-β) type I is restored in TGF-β-resistant hepatoma cells after expression of TGF-beta receptor type II cDNA. Proc. Natl. Acad. Sci. USA 1993, 90, 5359–5363, doi:10.1073/pnas.90.11.5359.
- Massague, J. The transforming growth factor-β family. Annu. Rev. Cell Biol. 1990, 6, 597–641, doi:10.1146/annurev.cb.06.110190.003121.
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, doi:10.1101/cshperspect.a021873.
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, doi:10.1126/scisignal.aav5183.
- Sun, P.D.; Davies, D.R. The cystine-knot growth-factor superfamily. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 269–291, doi:10.1146/annurev.bb.24.060195.001413.
- Batlle, E.; Massague, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940, doi:10.1016/j.immuni.2019.03.024.
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20112767.
- Levy, L.; Hill, C.S. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58, doi:10.1016/j.cytogfr.2005.09.009.
- Massague, J. TGF-β signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791, doi:10.1146/annurev.biochem.67.1.753.
- Lebrun, J.J.; Vale, W.W. Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Mol. Cell. Biol. 1997, 17, 1682–1691, doi:10.1128/mcb.17.3.1682.
- Yamashita, H.; ten Dijke, P.; Franzen, P.; Miyazono, K.; Heldin, C.H. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-β. J. Biol. Chem. 1994, 269, 20172–20178.
- Weis-Garcia, F.; Massague, J. Complementation between kinase-defective and activation-defective TGF-β receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. 1996, 15, 276–289.
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347, doi:10.1038/370341a0.
- Wieser, R.; Wrana, J.L.; Massague, J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995, 14, 2199–2208.
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, doi:10.1101/cshperspect.a022053.
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9, doi:10.1101/cshperspect.a022129.
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, doi:10.1101/cshperspect.a022061.
- David, C.J.; Massague, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435, doi:10.1038/s41580-018-0007-0.
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, doi:10.1101/cshperspect.a022137.
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012, 586, 1871–1884, doi:10.1016/j.febslet.2012.05.010.
- Iyengar, P.V. Regulation of Ubiquitin Enzymes in the TGF-β Pathway. Int. J. Mol. Sci. 2017, 18, doi:10.3390/ijms18040877.
- Tu, A.W.; Luo, K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J. Biol. Chem. 2007, 282, 21187–21196, doi:10.1074/jbc.M700085200.
- Simonsson, M.; Kanduri, M.; Gronroos, E.; Heldin, C.H.; Ericsson, J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J. Biol. Chem. 2006, 281, 39870–39880, doi:10.1074/jbc.M607868200.
- Kang, J.S.; Saunier, E.F.; Akhurst, R.J.; Derynck, R. The type I TGF-β receptor is covalently modified and regulated by sumoylation. Nat. Cell Biol. 2008, 10, 654–664, doi:10.1038/ncb1728.
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 2000, 6, 1365–1375, doi:10.1016/s1097-2765(00)00134-9.
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-β (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-β type I receptor. Biochem. J. 2005, 386, 461–470, doi:10.1042/BJ20040738.
- Wicks, S.J.; Haros, K.; Maillard, M.; Song, L.; Cohen, R.E.; Dijke, P.T.; Chantry, A. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-β signalling. Oncogene 2005, 24, 8080–8084, doi:10.1038/sj.onc.1208944.
- De Boeck, M.; Ten Dijke, P. Key role for ubiquitin protein modification in TGFβ signal transduction. Ups. J. Med. Sci. 2012, 117, 153–165, doi:10.3109/03009734.2012.654858.
- Imamura, T.; Oshima, Y.; Hikita, A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. J. Biochem. 2013, 154, 481–489, doi:10.1093/jb/mvt097.
- Liu, S.; de Boeck, M.; van Dam, H.; Ten Dijke, P. Regulation of the TGF-β pathway by deubiquitinases in cancer. Int. J. Biochem. Cell Biol. 2016, 76, 135–145, doi:10.1016/j.biocel.2016.05.001.
- Herhaus, L.; Sapkota, G.P. The emerging roles of deubiquitylating enzymes (DUBs) in the TGFβ and BMP pathways. Cell. Signal. 2014, 26, 2186–2192, doi:10.1016/j.cellsig.2014.06.012.
- Kim, S.Y.; Baek, K.H. TGF-β signaling pathway mediated by deubiquitinating enzymes. Cell. Mol. Life Sci. 2019, 76, 653–665, doi:10.1007/s00018-018-2949-y.
- Abdollah, S.; Macias-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685, doi:10.1074/jbc.272.44.27678.
- Souchelnytskyi, S.; Tamaki, K.; Engstrom, U.; Wernstedt, C.; ten Dijke, P.; Heldin, C.H. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J. Biol. Chem. 1997, 272, 28107–28115, doi:10.1074/jbc.272.44.28107.
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; Ten Dijke, P. Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors. Bone 2020, 138, 115472, doi:10.1016/j.bone.2020.115472.
- Lagna, G.; Hata, A.; Hemmati-Brivanlou, A.; Massague, J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature 1996, 383, 832–836, doi:10.1038/383832a0.
- Wu, R.Y.; Zhang, Y.; Feng, X.H.; Derynck, R. Heteromeric and homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol. Cell. Biol. 1997, 17, 2521–2528, doi:10.1128/mcb.17.5.2521.
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362, doi:10.1093/emboj/16.17.5353.
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, doi:10.1101/cshperspect.a022079.
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100, doi:10.1093/emboj/17.11.3091.
- Itoh, S.; ten Dijke, P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 2007, 19, 176–184, doi:10.1016/j.ceb.2007.02.015.
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, doi:10.1101/cshperspect.a022095.
- de Ceuninck van Capelle, C.; Spit, M.; Ten Dijke, P. Current perspectives on inhibitory SMAD7 in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 691–715, doi:10.1080/10409238.2020.1828260.
- Datta, P.K.; Moses, H.L. STRAP and Smad7 synergize in the inhibition of transforming growth factor β signaling. Mol. Cell. Biol. 2000, 20, 3157–3167, doi:10.1128/mcb.20.9.3157-3167.2000.
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L'Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-β/Smad signaling. Oncogene 2002, 21, 4879–4884, doi:10.1038/sj.onc.1205623.
- Lallemand, F.; Seo, S.R.; Ferrand, N.; Pessah, M.; L'Hoste, S.; Rawadi, G.; Roman-Roman, S.; Camonis, J.; Atfi, A. AIP4 restricts transforming growth factor-β signaling through a ubiquitination-independent mechanism. J. Biol. Chem. 2005, 280, 27645–27653, doi:10.1074/jbc.M500188200.
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700, doi:10.1016/s0092-8674(03)00432-x.
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584, doi:10.1242/jcs.02554.
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-β-induced gene expression. J. Biol. Chem. 1999, 274, 27161–27167, doi:10.1074/jbc.274.38.27161.
- Bakin, A.V.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell. Sci. 2002, 115, 3193–3206.
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002, 21, 3749–3759, doi:10.1093/emboj/cdf366.
- Sano, Y.; Harada, J.; Tashiro, S.; Gotoh-Mandeville, R.; Maekawa, T.; Ishii, S. ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-β signaling. J. Biol. Chem. 1999, 274, 8949–8957, doi:10.1074/jbc.274.13.8949.
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967, doi:10.1038/sj.emboj.7601818.
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620, doi:10.1016/j.tibs.2013.10.001.
- Edlund, S.; Landstrom, M.; Heldin, C.H.; Aspenstrom, P. Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914, doi:10.1091/mbc.01-08-0398.
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36, doi:10.1091/mbc.12.1.27.
- Shen, X.; Li, J.; Hu, P.P.; Waddell, D.; Zhang, J.; Wang, X.F. The activity of guanine exchange factor NET1 is essential for transforming growth factor-β-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 15362–15368, doi:10.1074/jbc.M009534200.
- Papadimitriou, E.; Kardassis, D.; Moustakas, A.; Stournaras, C. TGFβ-induced early activation of the small GTPase RhoA is Smad2/3-independent and involves Src and the guanine nucleotide exchange factor Vav2. Cell. Physiol. Biochem. 2011, 28, 229–238, doi:10.1159/000331734.
- Kim, S.J.; Im, Y.H.; Markowitz, S.D.; Bang, Y.J. Molecular mechanisms of inactivation of TGF-β receptors during carcinogenesis. Cytokine Growth Factor Rev. 2000, 11, 159–168, doi:10.1016/s1359-6101(99)00039-8.
- Li, J.M.; Nichols, M.A.; Chandrasekharan, S.; Xiong, Y.; Wang, X.F. Transforming growth factor β activates the promoter of cyclin-dependent kinase inhibitor p15INK4B through an Sp1 consensus site. J. Biol. Chem. 1995, 270, 26750–26753, doi:10.1074/jbc.270.45.26750.
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549, doi:10.1073/pnas.92.12.5545.
- Coffey, R.J., Jr.; Bascom, C.C.; Sipes, N.J.; Graves-Deal, R.; Weissman, B.E.; Moses, H.L. Selective inhibition of growth-related gene expression in murine keratinocytes by transforming growth factor β. Mol. Cell. Biol. 1988, 8, 3088–3093, doi:10.1128/mcb.8.8.3088.
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled receptor kinase-2 is a TGFβ-inducible antagonist of TGFβ signal transduction. EMBO J. 2005, 24, 3247–3258, doi:10.1038/sj.emboj.7600794.
- Kang, Y.; Chen, C.R.; Massague, J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 2003, 11, 915–926, doi:10.1016/s1097-2765(03)00109-6.
- Kowanetz, M.; Valcourt, U.; Bergstrom, R.; Heldin, C.H.; Moustakas, A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor β and bone morphogenetic protein. Mol. Cell. Biol. 2004, 24, 4241–4254, doi:10.1128/mcb.24.10.4241-4254.2004.
- Ohgushi, M.; Kuroki, S.; Fukamachi, H.; O'Reilly, L.A.; Kuida, K.; Strasser, A.; Yonehara, S. Transforming growth factor β-dependent sequential activation of Smad, Bim, and caspase-9 mediates physiological apoptosis in gastric epithelial cells. Mol. Cell. Biol. 2005, 25, 10017–10028, doi:10.1128/MCB.25.22.10017-10028.2005.
- Wildey, G.M.; Patil, S.; Howe, P.H. Smad3 potentiates transforming growth factor beta (TGFβ)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J. Biol. Chem. 2003, 278, 18069–18077, doi:10.1074/jbc.M211958200.
- Francis, J.M.; Heyworth, C.M.; Spooncer, E.; Pierce, A.; Dexter, T.M.; Whetton, A.D. Transforming growth factor-β1 induces apoptosis independently of p53 and selectively reduces expression of Bcl-2 in multipotent hematopoietic cells. J. Biol. Chem. 2000, 275, 39137–39145, doi:10.1074/jbc.M007212200.
- Saltzman, A.; Munro, R.; Searfoss, G.; Franks, C.; Jaye, M.; Ivashchenko, Y. Transforming growth factor-β-mediated apoptosis in the Ramos B-lymphoma cell line is accompanied by caspase activation and Bcl-XL downregulation. Exp. Cell Res. 1998, 242, 244–254, doi:10.1006/excr.1998.4096.
- Chipuk, J.E.; Bhat, M.; Hsing, A.Y.; Ma, J.; Danielpour, D. Bcl-xL blocks transforming growth factor-β1-induced apoptosis by inhibiting cytochrome c release and not by directly antagonizing Apaf-1-dependent caspase activation in prostate epithelial cells. J. Biol. Chem. 2001, 276, 26614–26621, doi:10.1074/jbc.M100913200.
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58, doi:10.1038/ncb731.
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978, doi:10.7150/ijbs.4564.
- Glick, A.B.; Weinberg, W.C.; Wu, I.H.; Quan, W.; Yuspa, S.H. Transforming growth factor β1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996, 56, 3645–3650.
- Katz, L.H.; Li, Y.; Chen, J.S.; Munoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760, doi:10.1517/14728222.2013.782287.
- Kirshner, J.; Jobling, M.F.; Pajares, M.J.; Ravani, S.A.; Glick, A.B.; Lavin, M.J.; Koslov, S.; Shiloh, Y.; Barcellos-Hoff, M.H. Inhibition of transforming growth factor-β1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 2006, 66, 10861–10869, doi:10.1158/0008-5472.CAN-06-2565.
- Bornstein, S.; White, R.; Malkoski, S.; Oka, M.; Han, G.; Cleaver, T.; Reh, D.; Andersen, P.; Gross, N.; Olson, S.; et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J. Clin. Investig. 2009, 119, 3408–3419, doi:10.1172/JCI38854.
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183, doi:10.1186/1479-5876-10-183.
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002, doi:10.1091/mbc.e04-08-0658.
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 205–218, doi:10.1002/path.2785.
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520, doi:10.1111/j.1349-7006.2007.00550.x.
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-β in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454, doi:10.1042/BST20160065.
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296, doi:10.1038/ncb1973.
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549, doi:10.1016/s1535-6108(03)00132-6.
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206, doi:10.1172/JCI3523.
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbi, A.; Attisano, L.; Bernabeu, C. Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535, doi:10.1074/jbc.M104536200.
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129, doi:10.1038/ng1001-117.
- Rossant, J.; Howard, L. Signaling pathways in vascular development. Annu. Rev. Cell Dev. Biol. 2002, 18, 541–573, doi:10.1146/annurev.cellbio.18.012502.105825.
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227, doi:10.1016/j.it.2010.04.002.
- Arteaga, C.L.; Hurd, S.D.; Winnier, A.R.; Johnson, M.D.; Fendly, B.M.; Forbes, J.T. Anti-transforming growth factor (TGF)- β antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-β interactions in human breast cancer progression. J. Clin. Investig. 1993, 92, 2569–2576, doi:10.1172/JCI116871.
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567, doi:10.1038/nri2808.


