 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diana Cimiotti | + 2533 word(s) | 2533 | 2021-01-19 03:46:52 | | | |
| 2 | Diana Cimiotti | -2 word(s) | 2531 | 2021-01-22 11:03:14 | | | | |
| 3 | Camila Xu | Meta information modification | 2531 | 2021-02-04 05:23:08 | | |
Video Upload Options
Cardiomyopathies are defined as cardiac diseases, in which the heart muscle is affected showing functional and structural defects.
1. Cardiomyopathies
According to the classification of cardiomyopathies as described by Elliott et al., 2008, they can be subdivided into RCM (restrictive cardiomyopathy), HCM (hypertrophic cardiomyopathy), DCM (dilated cardiomyopathy), ACM (arrhythmogenic cardiomyopathy) and unclassified cardiomyopathies as for example non-compaction cardiomyopathy (LVNC) (Figure 1). The causes of these cardiomyopathies may be genetic/familial or non-genetic and idiopathic.
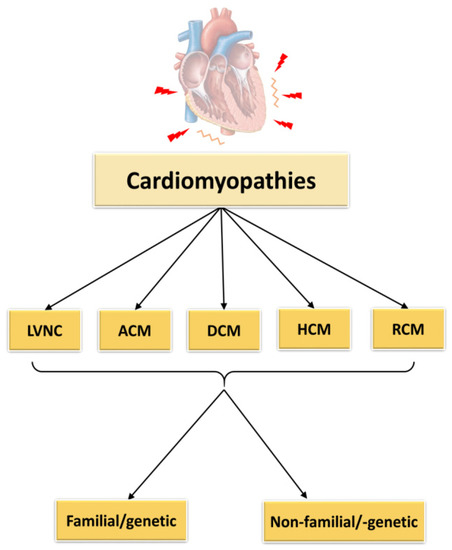
Figure 1. Classification of cardiomyopathies. LVNC = left ventricular non-compaction cardiomyopathy, ACM = arrhythmogenic cardiomyopathy, DCM = dilated cardiomyopathy, HCM = hypertrophic cardiomyopathy, RCM = restrictive cardiomyopathy.
1.1. Left Ventricular Non-Compaction Cardiomyopathy
LVNC seems to be the common cardiomyopathy type in the class of unclassified cardiomyopathies at least in children . The origin of the disease is thought to lie in an impaired embryonic development, leading to a sponge-like ventricle and dilation due to abnormal trabeculations. In several cases mutations have been identified for example in the genes encoding a member of the dystrophin–related protein family, tafazzin, a mitochondrial membrane protein involved in cardiolipin metabolism, in dystrobrevin, or in a gene encoding lamin, located in the nuclear envelope . But also sarcomeric genes as MYH7, ACTC, TNNT2, TPM1, ZASP are affected [1]. TNNT2 seems to be involved in cardiogenesis in the regulation of atrial septal growth and formation of trabeculae [2]. However, the mechanism of disease development still is obscure and it is not clear, if and how these mutations impair correct embryonic development of the heart. Clinically, LVNC is associated with left ventricular dysfunction and severe arrhythmia, sudden cardiac death, or embolic stroke due to an enhanced risk of thrombus formation within the trabeculaes.
1.2. Arrhythmogenic Cardiomyopathy
ACM has an estimated frequency in the general population of 1:100 to 1:5000. Since sudden cardiac death similar to hypertrophic cardiomyopathy may occur as the first manifestation of the disease, there might be an additional number of unreported cases of ACM in the general population [3]. The hallmark of ACM is the replacement of ventricular cardiomyocytes by fibrotic and fatty tissue, which progresses with time. This might affect the right or left ventricle, or both, and finally leads to electrical instability and systolic dysfunction [4][5]. Like the other cardiomyopathies, ACM is genetically heterogeneous. Thus, for example, variants of proteins of the nuclear envelope like transmembrane protein 43 or lamin A/C have been correlated to ACM. The latter seems to induce mainly right ventricular and bi-ventricular cardiomyopathy [6][7]. In addition, mutations in PLN encoding phospholamban have been identified to cause ACM. In the Netherlands, the phospholamban p.R14del mutation has been declared a founder mutation responsible for the disease in 10–15% of all ACM patients [8]. Other ACM target genes encode the cardiac sodium channel and the sarcomeric protein titin [9][10][11]. Several rare single amino acid replacements in titin have been identified in different families (p.T8031C, p.A18579I + p.M33291T, p.A19309S, p.P308471L, p.T2896I). p.T2896I is located in the conserved Ig10 domain within the spring region of titin [10]. Also, nonsense filamin C variants have been correlated to either DCM or ACM. Recently, a filamin C intronic mutation was described in three Jewish families leading to reduced filamin C transcripts as well as aberrant filamin C protein variants [12]. Interestingly, these variants did not show a mislocalization of proteins such as glycogen synthase kinase-3β or plakoglobin considered typical for ACM [13]. Mainly, ACM is caused by mutations in genes encoding structural proteins as desmosomal proteins like plakoglobin, desmoplakin, plakophillin etc., with increased risk of sudden cardiac death and left ventricular dysfunction [14]. Desmosomes link desmin to the extracellular matrix. Dysfunctional desmosomes not only affect cell-cell communication, but also lead to cell death. Such a loss of cardiomyocytes is compensated finally by substitution with fat and/or fibrous tissue instead of new cardiomyocytes, since the regeneration capacity of cardiomyocytes is extremely low [15]. The disease usually manifests in adults or during adolescence. It is very rarely diagnosed in children, probably because of lacking symptoms. However, an early diagnosis would help to postpone manifestation of a severe ACM [16][17][18].
1.3. Dilated Cardiomyopathy
One of the common cardiomyopathies is DCM, which is mainly characterized by left ventricle dilation, systolic dysfunction and high morbidity. Main causes for DCM are infections, inflammation or toxins [19]. Also infants might be affected showing either mild or strong symptoms at diagnosis, but the disease onset in childhood is generally correlated with a high mortality rate. In a Swedish study, only 8% of the children recovered within the 25-year follow-up period [20]. Most of them had to undergo a heart transplantation, or a ventricular assist device or pacemaker was implanted, or they died before any of these options could be applied. An American study revealed that boys were more affected than girls and that also the ethnic origin seemed to play a role in disease progression [21]. Less frequently, DCM may also be caused by genetic defects, though there might be a significant number of undetected cases. Thus, according to Burkett & Hershberger, idiopathic DCM to about 50% is due to mutations [22]. The inheritance mainly is autosomal dominant, but also could be recessive, X-linked or even mitochondrial [23]. More than 60 genes have been associated to familial DCM [24][25]. The target genes for example might encode the sarcomeric proteins titin, cardiac troponin T (cTnT) and C (cTnC), actin, myosin heavy chain (MHC) or ion channels as the voltage gated sodium channel subunit alpha, as well as structural proteins e.g. lamin, filamin C, desmin [13][15]. One of the most prominent genes affected in familial DCM is TTN (up to 30% of all familial DCM cases up to date) encoding titin, the elastic filament of the sarcomere [26]. Here, mostly truncation mutations have been observed in up to 25% of young DCM patients [27][28]. Most titin truncations in DCM patients occur in the A-band region of the sarcomere, whereby penetrance is clearly age dependent [29][30]. But also missense mutations have been described in TTN leading to similarly severe DCM as observed in patients with truncated titin. For example, Galan et al., 2020, recently showed that replacement of functional active cysteine residues in titin, whose oxidation affects titin stiffness and dynamics, leads to the development of DCM [31].
1.4. Hypertrophic Cardiomyopathy
The most frequent cardiomyopathy based on gene defects is HCM, whereby more than 1400 mutations in genes mainly encoding sarcomeric proteins have been identified up to date. Clinically, HCM is characterized by symmetrically or asymmetrically thickened heart walls, affecting in most cases the septum and/or the left ventricle. Diastolic dysfunction and a high risk of sudden cardiac death especially in young athletes are hallmarks in HCM. Histologically, cardiomyocytes appear enlarged and disarrayed and the cardiac muscle tissue shows fibrosis. In general, the disease manifestation is highly variable. Though there are also severe cases in young people, often the disease remains asymptomatic and thus undetected in the young [32][33][34]. The genes which are affected most in HCM patients are those encoding for myosin heavy chain (MHC) and cardiac myosin binding protein C (cMyBP-C). More than 50% of the reported HCM mutations have been detected in these two genes [35]. In MYH7 (cardiac gene of MHC) and most other genes encoding sarcomeric proteins, predominantly missense mutations are found leading to single amino acid replacements in the resulting protein. In MHC, mostly amino acid replacements in the actin binding domain or the ATPase domain have been identified, affecting force production [36]. In case of cMyBP-C, mainly truncated proteins are formed, leading to haploinsufficiency [37]. On the molecular level, when using isolated recombinant protein fragments of MHC variants in functional assays, MYH7 HCM mutations reduced force production. In contrast, in animal models and isolated variant MHCs enhanced contractility, i.e., increased and accelerated force production was observed [38]. The discrepancy of these observations may be due to effects of post-translational modifications or involvement of other proteins present in the more complex assay systems such as animal models and isolated whole cells or even myofibrils. Due to the complexity of the sarcomere and its interactions, reduced assay systems as reconstituted filaments are not able to reflect the situation in the sarcomere or even less in a cardiomyocyte or tissue; they just show dysfunction of the used proteins, but not necessarily the outcome in the cardiomyocyte or tissue. Thus, the enhancement of contractility fits to the generally observed increased Ca2+-sensitivity leading to enhanced activation at lower calcium concentrations than in healthy cardiac muscle [39][40]. Such a hypercontractility is known to lead to energy (ATP) depletion, but more importantly increase ADP- and decrease phosphocreatine levels, thereby affecting myosin cross bridges, force production and impairing re-extension [41][42].
1.5. Restrictive Cardiomyopathy
Restrictive cardiomyopathy (RCM) is a lethal, but rare disease which mostly is due to infiltration, and in a smaller percentage due to genetic disorders. In general, genetic RCM is characterized by near normal-sized left ventricle with enhanced stiffness and enlarged atria due to increased end-diastolic pressure in the ventricles. The disease is combined with an abnormal filling pattern and thus belongs to the diastolic diseases. Systolic function at least in the beginning of the disease is near normal but might be reduced at later stages of the disease. Sometimes also a mild hypertrophy is observed, making diagnostic distinction between RCM and HCM difficult [43].
The far most common cause of infiltrative diseases is amyloidosis that results from misfolding and deposition of proteins (amyloids) between the muscle fibers and/or within the walls of coronary arteries. The amyloids induce an enlargement of the heart walls giving an appearance of hypertrophy. However, the myofibers themselves are not affected as they are in HCM [44]. Two main types can be distinguished: the light chain (AL) and the transthyretin amyloidosis (ATTR). The latter type includes a hereditary sub-type caused by variants of the transthyretin protein, and a more common wild-type ATTR which is clearly age-related (“senile ATTR”) [45]. Similar as for DCM, the majority of idiopathic RCM cases are caused by gene defects, though up to date the knowledge on RCM genetics is still very poor [46][47]. In genetically based RCM, the inheritance usually is autosomal dominant. Genes with (non-infiltrative) RCM variants include also TNNI3, TNNT2, TNNC1, TPM1, TTN, MYH7, MYL2, MYBPC3, MPN, DES, FLNC, LMNA, BAG3 (Table 1) and are similar to those of DCM, HCM and LVNC [46][48][49]. Most mutations have been identified in genes encoding for sarcomeric proteins, some in sarcomere associated proteins like small heatshock proteins such as crystallin αB, or their binding partners such as BAG3 — proteins whose dysfunction potentially leads to the accumulation of aggregated proteins.
Several mutations in genes whose proteins are not directly involved in contractile function have been described in patients with RCM, among others desmin, filamin C and crystallin αB [50][51][52][53][54]. Usually, desmin mutations have been associated with DCM, however, a p.E413K mutation was found in a Polish family with a history of fatal heart diseases, in which 3 adult (30–60 year old) living members suffered of RCM [50]. Other family members also in young age suffered from heart disease and dies suddenly, but the diagnostic confirmation of RCM was not clear. The p.E413K desmin mutation is located in a conserved region involved in filament assembly which is different to the other mutations found in DCM patients. In patient muscle biopsies as well as in a cell culture model, desmin aggregates and disrupted Z-discs have been observed. Also, another desmin mutation which has been linked to RCM affects a splicing site within the DES gene and leads to disruption of the filamentous network of the cardiomyocytes [52]. In this case cardiac symptoms were diagnosed at the age of 46 in a Polish patient. More recently, a homozygous p.Y122H desmin mutation was identified in a RCM patient aged 19 [51]. This mutation is located within a region which is involved in the coiled coil formation of desmin dimers, and leads to abnormal cytoplasmic aggregation of desmin suggesting that this region may be a hotspot of cardiomyopathy-related mutations.
Pediatric RCM
Cardiomyopathies in children are overall rare, but often they are associated with a poor prognosis. The most common cardiomyopathies in children are DCM followed by HCM [21][55]. In children, genetically based RCM is seldom and accounts for less than 5% of the cardiomyopathy cases. Children with RCM show a rapid disease progression as well as a high mortality (50% survival within the first two years after diagnosis) [56][57]. Clinical characteristics are similar to RCM in adults, with diastolic dysfunction in absence of hypertrophy. In addition, pulmonary venous congestion, atrial fibrillation and SA block may occur, associated with an increased risk for arrhythmia and sudden cardiac death. [49][56]. Mogenson & Arbustini, 2009, suggested, that children with RCM exhibit a high risk for ischemia related events (infarcts, scarring, necrosis) even without signs of heart failure [58].
Several mutations have been detected in children with RCM affecting structural proteins, among which mutations in FLNC seem to be most prominent. Filamin C cross-links actin filaments and is located at costameres, Z-discs and intercalated discs. The first RCM mutations in FLNC were described by Brodehl et al., 2016, in two different Canadian families leading to single amino acid replacements in conserved immunoglobulin domains, p.S1624L and p.I2160F [59]. Tissues of patients with p.S1624L showed filamin C aggregates and disrupted Z-discs. Members of this family became diseased at an age <10 years. In the family with p.I2160F, no aggregates were detected and the onset of the disease occurred much later. More recently two de novo mutations in FLNC have been found in children diagnosed with RCM at the age of 1, 3 and 15 years for the p.A1186V mutation and 6 months for the p.A1183L mutation [53]. Both mutations cause abnormal filamin C localization, disruption of Z-disks as well as aggregation. Similarly, a p.P209L mutation in BAG3, identified in 2018 in an eight year old boy diagnosed with myofibrillar myopathy and RCM, also caused aggregation of BAG3 and desmin, Z-disc abnormalities as well as dysregulated autophagy [60]. Though only relatively few mutations have been thoroughly characterized so far, myofibrillar disarray and protein aggregation seem to be common features in many mutations analyzed, supporting the idea of an infiltrative pathomechanism of RCM.
Other targets for RCM mutations are genes encoding sarcomeric proteins. In this group the main target is TNNI3 encoding cardiac troponin I (cTnI), a sarcomeric regulatory protein [61]. TNNI3 mutations were found to be predominant in pediatric RCM in a Chinese study [62]. Here, as well, detailed analyses of the underlying mechanisms are scarce, most suggesting contractile abnormalities such as increased Ca2+-sensitivity and impaired relaxation, which also occur in HCM. Interestingly, there seems to be a high rate of de novo infantile RCM mutations in the TNNI3 gene, though a few de novo mutations have also been observed in TTN and MYH7 [46][63][64][58].
De novo mutations as disease causing mutations are not easy to identify, especially in case of missense mutations leading to a single amino acid replacement [65]. Several factors have to be considered, as for example the localization of the mutation in a disease gene, the conserved position of the amino acid replacement and the function of the resulting protein, etc. A number of de novo mutations have been identified in pediatric cardiomyopathies. They come along with a very fast disease progression and a poor prognosis. Pediatric RCM patients with de novo mutations frequently require a heart transplantation shortly after diagnosis to prevent premature death. Only a few of the known mutations, however, have been investigated on the mechanistic level.
References
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Głowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Kłyś, J.; Venner, E.; et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients with Left Ventricular Noncompaction. Circ. Car-diovasc. Genet. 2017, 10, doi:10.1161/CIRCGENETICS.117.001763.
- England, J.; Pang, K.L.; Parnall, M.; Haig, M.I.; Loughna, S. Cardiac Troponin T Is Necessary for Normal Development in the Embryonic Chick Heart. J. Anat. 2016, 229, 436–449, doi:10.1111/joa.12486.
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ven-tricular Dysplasia/ Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446, doi:10.1161/CIRCGENETICS.114.001003.
- Beffagna, G.; Zorzi, A.; Pilichou, K.; Perazzolo Marra, M.; Rigato, I.; Corrado, D.; Migliore, F.; Rampazzo, A.; Bauce, B.; Bas-so, C.; et al. Arrhythmogenic Cardiomyopathy. Eur. Heart J. 2020, 22, 1147–1148, doi:10.1093/eurheartj/ehaa719.
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 785–802, doi:10.1161/CIRCRESAHA.117.309345.
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadano-va, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Is a Fully Penetrant, Lethal Arrhythmic Disorder Caused by a Missense Mutation in the TMEM43 Gene. Am. J. Hum. Genet. 2008, 82, 809–821, doi:10.1016/j.ajhg.2008.01.010.
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C Gene Mimic Arrhythmogenic Right Ventricular Cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136, doi:10.1093/eurheartj/ehr451.
- Van Rijsingen, I.A.W.; Van Der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.H.; Zwinderman, A.H.; Pinto, Y.M.; Lekanne Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in Phospholamban R14del Carriers Results of a Large Multicentre Cohort Study. Circ. Cardiovasc. Genet. 2014, 7, 455–465, doi:10.1161/CIRCGENETICS.113.000374.
- Veerman, C.C.; Wilde, A.A.M.; Lodder, E.M. The Cardiac Sodium Channel Gene SCN5A and Its Gene Product NaV1.5: Role in Physiology and Pathophysiology. Gene 2015, 573, 177–187, doi:10.1016/j.gene.2015.08.062.
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic Variation in Titin in Arrhythmogenic Right Ventricular Cardiomyopathy-Overlap Syndromes. Circulation 2011, 124, 876–885, doi:10.1161/CIRCULATIONAHA.110.005405.
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and Desmosomal Genes in the Natural History of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Med. Genet. 2014, 51, 669–676, doi:10.1136/jmedgenet-2014-102591.
- Oz, S.; Yonath, H.; Visochyk, L.; Ofek, E.; Landa, N.; Reznik-Wolf, H.; Ortiz-Genga, M.; Monserrat, L.; Ben-Gal, T.; Goitein, O.; et al. Reduction in Filamin C Transcript Is Associated with Arrhythmogenic Cardiomyopathy in Ashkenazi Jews. Int. J. Cardiol. 2020, 317, 133–138, doi:10.1016/j.ijcard.2020.04.005.
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C Variants Are Associated with a Distinctive Clinical and Immunohistochemical Arrhythmogenic Cardiomyopathy Phenotype. Int. J. Cardiol. 2020, 307, 101–108, doi:10.1016/j.ijcard.2019.09.048.
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973, doi:10.1086/521633.
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heide, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102, doi:10.1126/science.1164680.
- Hamilton, R.M.; Fidler, L. Right Ventricular Cardiomyopathy in the Young: An Emerging Challenge. Hear. Rhythm 2009, 6, 571–575, doi:10.1016/j.hrthm.2009.01.026.
- Kriebel, T.; Korte, T.; Kandolf, R.; Jux, C.; Windhagen-Mahnert, B.; Bökenkamp, R.; Bertram, H.; Paul, T. Arrhythmogene Rechtsventrikuläre Dysplasie/Kardiomyopathie -Diagnostik Im Kindesalter. Z. Kardiol. 2003, 92, 418–424, doi:10.1007/s00392-003-0937-0.
- Dungan, W.T.; Garson, A.; Gillette, P.C. Arrhythmogenic Right Ventricular Dysplasia: A Cause of Ventricular Tachycardia in Children with Apparently Normal Hearts. Am. Heart J. 1981, 102, 745–750, doi:10.1016/0002-8703(81)90101-0.
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a Revised Definition of Dilated Cardiomyopathy, Hypokinetic Non-Dilated Cardiomyopathy, and Its Implications for Clinical Practice: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2016, 37, 1850–1858, doi:10.1093/eurheartj/ehv727.
- Fadl, S.; Wåhlander, H.; Fall, K.; Cao, Y.; Sunnegårdh, J. The Highest Mortality Rates in Childhood Dilated Cardiomyopathy Occur during the First Year after Diagnosis. Acta Paediatr. Int. J. Paediatr. 2018, 107, 672–677, doi:10.1111/apa.14183.
- Lipshultz, S.E.; Cochran, T.R.; Briston, D.A.; Brown, S.R.; Sambatakos, P.J.; Miller, T.L.; Carrillo, A.A.; Corcia, L.; Sanchez, J.E.; Diamond, M.B.; et al. Pediatric Cardiomyopathies: Causes, Epidemiology, Clinical Course, Preventive Strategies and Therapies. Future Cardiol. 2013, 9, 817–848, doi:10.2217/fca.13.66.
- Burkett, E.L.; Hershberger, R.E. Clinical and Genetic Issues in Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 969–981, doi:10.1016/j.jacc.2004.11.066.
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547, doi:10.1038/nrcardio.2013.105.
- Pérez-Serra, A.; Toro, R.; Sarquella-Brugada, G.; de Gonzalo-Calvo, D.; Cesar, S.; Carro, E.; Llorente-Cortes, V.; Iglesias, A.; Brugada, J.; Brugada, R.; et al. Genetic Basis of Dilated Cardiomyopathy. Int. J. Cardiol. 2016, 224, 461–472, doi:10.1016/j.ijcard.2016.09.068.
- Li, C.-J.; Chen, C.-S.; Yiang, G.-T.; Tsai, A.P.-Y.; Liao, W.-T.; Wu, M.-Y. Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies. J. Clin. Med. 2019, 8, 520, doi:10.3390/jcm8040520.
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the Clinical Genetics of Human Dilated Cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135, doi:10.1093/eurheartj/ehu301.
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin Mutations in IPS Cells Define Sarcomere Insufficiency as a Cause of Dilated Cardiomyopathy. Science 2015, 349, 982–986, doi:10.1126/science.aaa5458.
- Franaszczyk, M.; Chmielewski, P.; Truszkowska, G.; Stawinski, P.; Michalak, E.; Rydzanicz, M.; Sobieszczanska-Malek, M.; Pollak, A.; Szczygieł, J.; Kosinska, J.; et al. Titin Truncating Variants in Dilated Cardiomyopathy -Prevalence and Geno-type-Phenotype Correlations. PLoS ONE 2017, 12, e0169007, doi:10.1371/journal.pone.0169007.
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.G.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, doi:10.3389/fcvm.2016.00021.
- Jansweijer, J.A.; Nieuwhof, K.; Russo, F.; Hoorntje, E.T.; Jongbloed, J.D.H.; Lekanne Deprez, R.H.; Postma, A.V.; Bronk, M.; van Rijsingen, I.A.W.; de Haij, S.; et al. Truncating Titin Mutations Are Associated with a Mild and Treatable Form of Dilat-ed Cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 512–521, doi:10.1002/ejhf.673.
- Herrero Galán, E.; Dominguez, F.; Martinez-Martin, I.; Sanchez-Gonzalez, C.; Vicente, N.; Lalaguna, L.; Bonzon-Kulichenko, E.; Calvo, E.; Gonzalez-Lopez, E.; Cobo-Marcos, M.; et al. Conserved Cysteines in Titin Sustain the Mechanical Function of Cardiomyocytes. bioRxiv 2020, doi:10.1101/2020.09.05.282913.
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Haas, T.S.; Chan, R.H.M.; Udelson, J.E.; Garberich, R.F.; Lesser, J.R.; Appelbaum, E.; Manning, W.J.; et al. Risk Stratification and Outcome of Patients with Hypertrophic Cardiomyopathy ≥60 Years of Age. Circulation 2013, 127, 585–593, doi:10.1161/CIRCULATIONAHA.112.136085.
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Link, M.S.; Lesser, J.R.; Chan, R.H.M.; Garberich, R.F.; Udelson, J.E.; Maron, M.S. Hyper-trophic Cardiomyopathy in Adulthood Associated with Low Cardiovascular Mortality with Contemporary Management Strategies. J. Am. Coll. Cardiol. 2015, 65, 1915–1928, doi:10.1016/j.jacc.2015.02.061.
- Aro, A.L.; Nair, S.G.; Reinier, K.; Jayaraman, R.; Stecker, E.C.; Uy-Evanado, A.; Rusinaru, C.; Jui, J.; Chugh, S.S. Population Burden of Sudden Death Associated with Hypertrophic Cardiomyopathy. Circulation 2017, 136, 1665–1667, doi:10.1161/CIRCULATIONAHA.117.030616.
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Manage-ment. Biomolecules 2019, 9, 878, doi:10.3390/biom9120878.
- Marian, A.J.; Wu, Y.; Lim, D.S.; McCluggage, M.; Youker, K.; Yu, Q.T.; Brugada, R.; DeMayo, F.; Quinones, M.; Roberts, R. A Transgenic Rabbit Model for Human Hypertrophic Cardiomyopathy. J. Clin. Investig. 1999, 104, 1683–1692, doi:10.1172/JCI7956.
- Van Dijk, S.J.; Dooijes, D.; Dos Remedios, C.; Michels, M.; Lamers, J.M.J.; Winegrad, S.; Schlossarek, S.; Carrier, L.; Cate, F.J.T.; Stienen, G.J.M.; et al. Cardiac Myosin-Binding Protein C Mutations and Hypertrophic Cardiomyopathy. Circulation 2009, 119, 1473–1483, doi:10.1161/CIRCULATIONAHA.108.838672.
- Seidman, C.E.; Seidman, J.G. Identifying Sarcomere Gene Mutations in Hypertrophic Cardiomyopathy: A Personal History. Circ. Res. 2011, 108, 743–750, doi:10.1161/CIRCRESAHA.110.223834.
- Schober, T.; Huke, S.; Venkataraman, R.; Gryshchenko, O.; Kryshtal, D.; Hwang, H.S.; Baudenbacher, F.J.; Knollmann, B.C. Myofilament Ca Sensitization Increases Cytosolic Ca Binding Affinity, Alters Intracellular Ca Homeostasis, and Causes Pause-Dependent Ca-Triggered Arrhythmia. Circ. Res. 2012, 111, 170–179, doi:10.1161/CIRCRESAHA.112.270041.
- Knollmann, B.C.; Kirchhof, P.; Sirenko, S.G.; Degen, H.; Greene, A.E.; Schober, T.; Mackow, J.C.; Fabritz, L.; Potter, J.D.; Mo-rad, M. Familial Hypertrophic Cardiomyopathy-Linked Mutant Troponin T Causes Stress-Induced Ventricular Tachycardia and Ca2+-Dependent Action Potential Remodeling. Circ. Res. 2003, 92, 428–436, doi:10.1161/01.RES.0000059562.91384.1A.
- Sequeira, V.; Najafi, A.; Wijnker, P.J.M.; Dos Remedios, C.G.; Michels, M.; Kuster, D.W.D.; Van Der Velden, J. ADP-Stimulated Contraction: A Predictor of Thin-Filament Activation in Cardiac Disease. Proc. Natl. Acad. Sci. USA 2015, 112, E7003–E7012, doi:10.1073/pnas.1513843112.
- Sequeira, V.; Bertero, E.; Maack, C. Energetic Drain Driving Hypertrophic Cardiomyopathy. FEBS Lett. 2019, 593, 1616–1626, doi:10.1002/1873-3468.13496.
- Tariq, M. Importance of Genetic Evaluation and Testing in Pediatric Cardiomyopathy. World J. Cardiol. 2014, 6, 1156, doi:10.4330/wjc.v6.i11.1156.
- Nihoyannopoulos, P.; Dawson, D. Restrictive Cardiomyopathies. Eur. J. Echocardiogr. 2009, 10, doi:10.1093/ejechocard/jep156.
- Yamamoto, H.; Yokochi, T. Transthyretin Cardiac Amyloidosis: An Update on Diagnosis and Treatment. ESC Hear. Fail. 2019, 6, 1128–1139, doi:10.1002/ehf2.12518.
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic Re-strictive Cardiomyopathy Is Part of the Clinical Expression of Cardiac Troponin I Mutations. J. Clin. Investig. 2003, 111, 209–216, doi:10.1172/JCI16336.
- Gallego-Delgado, M.; Delgado, J.F.; Brossa-Loidi, V.; Palomo, J.; Marzoa-Rivas, R.; Perez-Villa, F.; Salazar-Mendiguchía, J.; Ruiz-Cano, M.J.; Gonzalez-Lopez, E.; Padron-Barthe, L.; et al. Idiopathic Restrictive Cardiomyopathy Is Primarily a Genetic Disease. J. Am. Coll. Cardiol. 2016, 67, 3021–3023.
- Kaski, J.P.; Syrris, P.; Burch, M.; Tomé Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic Restrictive Cardiomyopathy in Children Is Caused by Mutations in Cardiac Sarcomere Pro-tein Genes. Heart 2008, 94, 1478–1484, doi:10.1136/hrt.2007.134684.
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS ONE 2016, 11, e0163362, doi:10.1371/journal.pone.0163362.
- Pruszczyk, P.; Kostera-Pruszczyk, A.; Shatunov, A.; Goudeau, B.; Dramiñska, A.; Takeda, K.; Sambuughin, N.; Vicart, P.; Strelkov, S.V.; Goldfarb, L.G.; et al. Restrictive Cardiomyopathy with Atrioventricular Conduction Block Resulting from a Desmin Mutation. Int. J. Cardiol. 2007, 117, 244–253, doi:10.1016/j.ijcard.2006.05.019.
- Brodehl, A.; Hakimi, S.A.P.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy Is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918, doi:10.3390/genes10110918.
- Ojrzyńska, N.; Bilińska, Z.T.; Franaszczyk, M.; Płoski, R.; Grzybowski, J. Restrictive Cardiomyopathy Due to Novel Desmin Gene Mutation. Kardiol. Pol. 2017, 75, 723.
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De Novo Mutations in FLNC Leading to Early-Onset Restrictive Cardiomyopathy and Congenital Myo-pathy. Hum. Mutat. 2018, 39, 1161–1172, doi:10.1002/humu.23559.
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The Novel ΑB-Crystallin (CRYAB) Mutation p.D109G Causes Restrictive Cardiomyopathy. Hum. Mutat. 2017, 38, 947–952, doi:10.1002/humu.23248.
- Ranthe, M.F.; Carstensen, L.; Øyen, N.; Jensen, M.K.; Axelsson, A.; Wohlfahrt, J.; Melbye, M.; Bundgaard, H.; Boyd, H.A. Risk of Cardiomyopathy in Younger Persons with a Family History of Death from Cardiomyopathy: A Nationwide Family Study in a Cohort of 3.9 Million Persons. Circulation 2015, 132, 1013–1019, doi:10.1161/CIRCULATIONAHA.114.013478.
- Wittekind, S.G.; Ryan, T.D.; Gao, Z.; Zafar, F.; Czosek, R.J.; Chin, C.W.; Jefferies, J.L. Contemporary Outcomes of Pediatric Restrictive Cardiomyopathy: A Single-Center Experience. Pediatr. Cardiol. 2019, 40, 694–704, doi:10.1007/s00246-018-2043-0.
- Webber, S.A.; Lipshultz, S.E.; Sleeper, L.A.; Lu, M.; Wilkinson, J.D.; Addonizio, L.J.; Canter, C.E.; Colan, S.D.; Everitt, M.D.; Jefferies, J.L.; et al. Outcomes of Restrictive Cardiomyopathy in Childhood and the Influence of Phenotype: A Report from the Pediatric Cardiomyopathy Registry. Circulation 2012, 126, 1237–1244, doi:10.1161/CIRCULATIONAHA.112.104638.
- Mogensen, J.; Arbustini, E. Restrictive Cardiomyopathy. Curr. Opin. Cardiol. 2009, 24, 214–220, doi:10.1097/HCO.0b013e32832a1d2e.
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; Mckinnon, M.L.; Mcgilli-vray, B.; Alvarez, N.; et al. Mutations in FLNC Are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279, doi:10.1002/humu.22942.
- Schänzer, A.; Rupp, S.; Gräf, S.; Zengeler, D.; Jux, C.; Akintürk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated Autophagy in Restrictive Cardiomyopathy Due to Pro209Leu Mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399, doi:10.1016/j.ymgme.2018.01.001.
- Mogensen, J.; Hey, T.; Lambrecht, S. A Systematic Review of Phenotypic Features Associated With Cardiac Troponin I Mu-tations in Hereditary Cardiomyopathies. Can. J. Cardiol. 2015, 31, 1377–1385, doi:10.1016/j.cjca.2015.06.015.
- Ding, W.H.; Han, L.; Xiao, Y.Y.; Mo, Y.; Yang, J.; Wang, X.F.; Jin, M. Role of Whole-Exome Sequencing in Phenotype Classi-fication and Clinical Treatment of Pediatric Restrictive Cardiomyopathy. Chin. Med. J. 2017, 130, 2823–2828, doi:10.4103/0366-6999.219150.
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin Mutation in Familial Restrictive Cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30, doi:10.1016/j.ijcard.2013.11.037.
- Karam, S.; Raboisson, M.J.; Ducreux, C.; Chalabreysse, L.; Millat, G.; Bozio, A.; Bouvagnet, P. A de Novo Mutation of the Beta Cardiac Myosin Heavy Chain Gene in an Infantile Restrictive Cardiomyopathy. Congenit. Heart Dis. 2008, 3, 138–143, doi:10.1111/j.1747-0803.2008.00165.x.
- Veltman, J.A.; Brunner, H.G. De Novo Mutations in Human Genetic Disease. Nat. Rev. Genet. 2012, 13, 565–575.
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin Mutation in Familial Restrictive Cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30, doi:10.1016/j.ijcard.2013.11.037.
- Karam, S.; Raboisson, M.J.; Ducreux, C.; Chalabreysse, L.; Millat, G.; Bozio, A.; Bouvagnet, P. A de Novo Mutation of the Beta Cardiac Myosin Heavy Chain Gene in an Infantile Restrictive Cardiomyopathy. Congenit. Heart Dis. 2008, 3, 138–143, doi:10.1111/j.1747-0803.2008.00165.x.
- Veltman, J.A.; Brunner, H.G. De Novo Mutations in Human Genetic Disease. Nat. Rev. Genet. 2012, 13, 565–575.


