 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rishi Man Chugh | + 1677 word(s) | 1677 | 2021-01-14 03:53:40 | | | |
| 2 | Bruce Ren | -20 word(s) | 1657 | 2021-01-22 03:46:54 | | |
Video Upload Options
The new strain of coronavirus (severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2)) emerged in 2019 and hence is often referred to as coronavirus disease 2019 (COVID-19). This disease causes hypoxic respiratory failure and acute respiratory distress syn-drome (ARDS), and is considered as the cause of a global pandemic. Very limited reports in addi-tion to ex vivo model systems are available to understand the mechanism of action of this virus, which can be used for testing of any drug efficacy against virus infectivity. COVID-19 induces tissue stem cell loss, resulting inhibition of epithelial repair followed by inflammatory fibrotic con-sequences. Development of clinically relevant models is important to examine the impact of the COVID-19 virus in tissue stem cells among different organs.
1. Introduction
Coronaviruses are a large group of viruses that can cause serious complications in animals and humans. There are seven classes of coronaviruses that infect people, however, three of these can cause serious, or lethal outcomes in humans. These include severe acute respiratory syndrome or SARS coronavirus (SARS-CoV); Middle East respiratory syndrome (MERS) (MERS-CoV); and, most recently, the new coronavirus severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), which has resulted in a pandemic that has infected more than 87 million people and approaching 1. 9 million deaths worldwide as of January 05, 2021 (https://www.worldometers.info/coronavirus/), a statistic that is growing daily. Coronaviruses are well known as the common cause of upper respiratory symptoms such as a dry cough, sinusitis, loss of taste and smell, and labored breathing; however, for SARS-CoV-2, a variety of other new and unusual symptoms have also been recognized in both humans [1] and in animal models. This new virus strain emerged in 2019 and, hence, as mentioned, is referred to as COVID-19.
COVID-19 significantly depletes tissue resident stem cell population [2], resulting in impaired tissue regeneration and repair. Moreover, loss of stem progenitor cells triggers the inflammatory and later fibrotic consequences [3][4][5][6]. Therefore, mitigation of tissue stem cell loss should be an effective therapeutic strategy against COVID-19 pathogenesis.
2. Experimental Model System for Understanding COVID-19 Pathogenesis
The bulk of our knowledge about the pathogenesis of COVID-19 in humans is based on available clinical trial data and case studies since the outbreak, as well as some preclinical and cell-based testing. The preclinical models available include non-human primates [7] and murine models that express human ACE2 in genetically modified mice such as the K18-hACE2 mice [8], among other tools [9][10], as well as genetically modified virus to recognize murine ACEII [11]. However, there are very limited reports available on a suitable ex vivo model system that can be used to understand the mechanism of action of the virus and can also be used for testing of any drug efficacy against virus infectivity. Tissue-specific stem cell-derived organoid systems could be a better model to understand the effect of COVID-19 on stem cells in the human body (Figure 1).
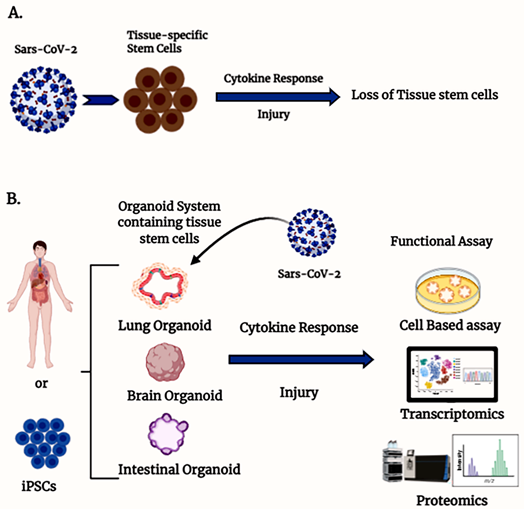
Figure 1. Organoids containing tissue stem cells are an ex vivo model to study severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) infection. (A) Schematic diagram illustrating the involvement of SARS-CoV-2-mediated stem cell loss. (B) Different organ-specific stem cell-derived organoid models to study SARS-CoV-2 infection.
3. Tissue-Specific Stem Cells and COVID Pathogenesis
There are several areas to study that are devoted to the antiviral approach by inhibiting replication (Remdesivir,), building immunity (vaccines), and their impact on the remediation of illness caused by the virus such as acute respiratory distress syndrome (ARDS). Antiviral therapy with Remdesivir has shown promise for reducing recovery time but is not be sufficient to inhibit lethal consequences from infection [12]. Antibody therapies also look highly encouraging if used early in the infection cycle [13], as do vaccines to reduce the spread and severity of the pandemic. Nevertheless, despite these advances, there is legitimate concern that COVID-19 may be present at some level in the population, albeit at a reduced rate, for a long period of time, and for those who contract the virus, long-term or permanent tissue damage is a real possibility in a percentage of cases worldwide.
Previous reports have indicated that in advanced COVID-19 cases, patients possess structural damage in lung epithelium [14][15] and COVID-19 significantly depletes the resident stem cell population [16]. The major pathological outcome due to COVID-19 is the damage to epithelial cells. It is important to note that this is not unique to COVID-19 as it has long been reported that SARS-CoV and H1N1 both propagate within type II cells where a large number of viral particles are released, and the cells undergo apoptosis [17]. These type II cells are presumed to function as progenitor cells that repair the injured alveolar epithelium [18][19][20]. Moreover, damaged epithelial cells also become a major source of inflammatory cytokines that not only can contribute to further damage to the tissue but have systemic and lethal effects as well [21]. Restitution and activation of pulmonary epithelial progenitor cells is critical to inhibit acute inflammation and suppression of pneumonitis/fibrosis (often referred to as ground glass in radiographic images). Loss of these progenitor cells by pathogenic or genotoxic stress impairs the regenerative process, resulting in a reduction in number of healthy epithelial cells, which eventually creates empty space for proliferation and repopulation of newly recruited inflammatory cells [22][23][24]. Moreover, damaged lung epithelial cells release inflammatory paracrine signals to promote recruitment of inflammatory cells. This is also well characterized in early studies with chemical injury as a model to demonstrate that loss of pulmonary stem progenitor cells triggers the inflammatory and later fibrotic consequences. Considering the extensive epithelial damage from COVID-19 virus infiltrate and the impact on lung stem/progenitor populations, further research is warranted and needed to gain a more complete understanding of the pathophysiology of COVID-19 infection. Mitigation of resident lung stem cells may be a key approach to minimize lung damage along with reduction in inflammation and fibrosis. It should also be considered that lungs from recovered COVID-19-infected patients may not regain full structural and functional integrity in severe cases since the repair or rebuilding capacity primarily depends on existing stem/progenitor populations. Early reports indicate lung epithelial stem cells may express SARS-CoV-2 entry factors higher than previously thought [25][26]. Lung contains functionally distinct candidate stem/progenitor cells such as basal cells [27], club cells [28][29], bronchoalveolar stem cells (BASCs) [30], and type II pneumocytes [31] involved in repair and regeneration of injured lungs. In addition to type II pneumocytes, several studies have revealed that a subset of murine and human Oct4+ pulmonary stem cells expressing ACE2 are the prime target of SARS-CoV infection [32][33], which leads to damage and loss of these cells [33].
As mentioned before, virulent forms of influenza viruses can infect various cell populations in the murine lung, but also display a strong tropism to an epithelial progenitor population defined by the signature EpCamhighCD24lowintegrin (α6β4)highCD200+expression. Three-dimensional organoid cultures derived from these epithelial stem/progenitor cells (EpiSPC), and in vivo infection models including transgenic mice, have shown that their enlargement, barrier regeneration, and outcome after virus-induced injury are highly dependent on Fgfr2b signaling. Importantly, virus-infected epithelial progenitor populations exhibited severely impaired renewal capacity due to virus-induced blockade of β-catenin-dependent Fgfr2b signaling, as evidenced by a loss of alveolar tissue repair capacity after intrapulmonary EpiSPC transplantation in vivo [34]. Wnt signaling is essential for lung epithelial stem cells repair and regeneration. The Wnt signaling pathway was downregulated in both in vivo-infected alveolar epithelial cells and in vitro-infected human lung epithelial A549 cells [35]. These results suggest that the influenza viruses may affect the host lung repair by regulating Wnt/β-catenin signaling. β- and γ-catenin regulate the innate cellular immune response to viruses by activating virus-dependent induction of the IFNB1 and downstream genes. Virulent viruses can suppress β-catenin-dependent transcription by misusing the RIG-I/NF-κB signaling cascade that is induced in the course of infection by viral RNA [36], and we hypothesize that COVID-19 is similar to other viruses in this regard [37]. Therefore, activation of Wnt/β-catenin signaling could be a major therapeutic intervention in the context of viral infection [38] if implemented early in the infectious lifecycle (Figure 2) where the immediate check on viral spread can happen before the adaptive immune response has time to develop several days after infection. More specifically, type I interferons are a critical part of our innate immune defense as they induce an array of proteins that interfere with virus replication in order to restrict and limit viral spread from cell to cell [39] in that early window before the adaptive immune response can even take effect. Viral suppression of this system may lead to unchecked and rapid spread, reaching very high viral loads in the lung and tissues. This, in turn would improve the chances of aerosolization and communication along with extensive tissue damage as the adaptive immune system takes over. While interferons have been used to treat COVID-19 with little success [40], biologically, its expression is timed as an immediate and early response rather than very late advanced disease where clinical trials have focused.
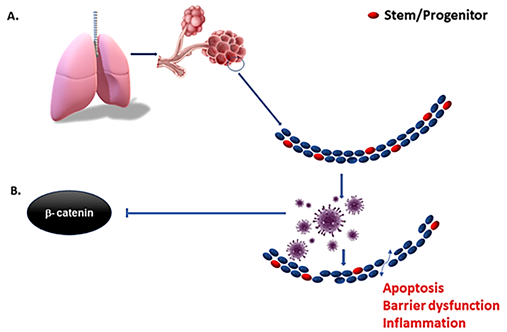
Figure 2. Early inhibition of interferons by SARS-CoV-2 and other viruses serve to suppress the innate immune response, resulting in rapid increases in cellular infection and spread before the adaptive immune response can develop. The depletion of resident stem cells by more virulent forms of viruses impedes the regenerative capacity of the tissue, and in turn increases the inflammatory context (A). Virulent forms of influenza suppress β-catenin nuclear localization (B) and downstream expression of interferons. Means of mitigating suppression of interferon and the innate immune response in the early phase of infection may reduce viral spread and preserve resident stem cells in the tissue of interest.
In COVID-positive patients, symptoms are also noted in multiple other organs, most notably the gastrointestinal tract and the kidney. Organoid-based studies demonstrated that SARS-CoV-2 could damage stem cells in these organs. However, the effects of SARS-CoV-2 in different type of stem cells such as intestinal quiescent stem cell populations vs. Lgr5+ active stem cell population is not known. Similarly, effect of SARS-CoV-2 on pancreatic and liver stem cells are predicted but further details are yet to be revealed.
References
- Marshall, M. The lasting misery of coronavirus long-haulers. Nature 2020, 585, 339–341, doi:10.1038/d41586-020-02598-6.
- Basiri, A.; Pazhouhnia, Z.; Beheshtizadeh, N.; Hoseinpour, M.; Saghazadeh, A.; Rezaei, N. Regenerative Medicine in COVID-19 Treatment: Real Opportunities and Range of Promises. Stem Cell Rev. Rep. 2020, 2020, 1–13, doi:10.1007/s12015-020-09994-5.
- Huang, K.; Kang, X.; Wang, X.; Wu, S.; Xiao, J.; Li, Z.; Wu, X.; Zhang, W. Conversion of bone marrow mesenchymal stem cells into type II alveolar epithelial cells reduces pulmonary fibrosis by decreasing oxidative stress in rats. Mol. Med. Rep. 2015, 11, 1685–1692, doi:10.3892/mmr.2014.2981.
- Nicolay, N.H.; Lopez Perez, R.; Rühle, A.; Trinh, T.; Sisombath, S.; Weber, K.-J.; Ho, A.D.; Debus, J.; Saffrich, R.; Huber, P.E. Mesenchymal stem cells maintain their defining stem cell characteristics after treatment with cisplatin. Sci. Rep. 2016, 6, 20035, doi:10.1038/srep20035.
- Nicolay, N.H.; Rühle, A.; Perez, R.L.; Trinh, T.; Sisombath, S.; Weber, K.J.; Ho, A.D.; Debus, J.; Saffrich, R.; Huber, P.E. Mesenchymal stem cells are sensitive to bleomycin treatment. Sci. Rep. 2016, 6, 26645, doi:10.1038/srep26645.
- Chen, X.; Wu, Y.; Wang, Y.; Chen, L.; Zheng, W.; Zhou, S.; Xu, H.; Li, Y.; Yuan, L.; Xiang, C. Human menstrual blood-derived stem cells mitigate bleomycin-induced pulmonary fibrosis through anti-apoptosis and anti-inflammatory effects. Stem Cell Res. Ther. 2020, 11, 1–19, doi:10.1186/s13287-020-01926-x.
- Lu, S.; Zhao, Y.; Yu, W.; Yang, Y.; Gao, J.; Wang, J.; Kuang, D.; Yang, M.; Yang, J.; Ma, C.; et al. Comparison of nonhuman primates identified the suitable model for COVID-19. Signal Transduct. Target. Ther. 2020, 5, 1–9, doi:10.1038/s41392-020-00269-6.
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal Infection of K18-hACE2 Mice Infected with Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 813–821, doi:10.1128/jvi.02012-06.
- Huang, C.; Peters, C.J.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus Accessory Protein 6 Is a Viri-on-Associated Protein and Is Released from 6 Protein-Expressing Cells. J. Virol. 2007, 81, 5423–5426, doi:10.1128/jvi.02307-06.
- Muñoz-Fontela, C.; Dowling, W.E.; Funnell, S.G.P.; Gsell, P.-S.; Riveros-Balta, A.X.; Albrecht, R.A.; Andersen, H.; Baric, R.S.; Carroll, M.W.; Cavaleri, M.; et al. Animal models for COVID-19. Nature 2020, 586, 509–515, doi:10.1038/s41586-020-2787-6.
- Dinnon, K.H., 3rd; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L., Jr.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566, doi:10.1038/s41586-020-2708-8.
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826, doi:10.1056/NEJMoa2007764.
- DeFrancesco, L. COVID-19 antibodies on trial. Nat Biotechnol 2020, 38, 1242–1252, doi.org/10.1038/s41587-020-0732-8.
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140, doi:10.1016/s1473-3099(20)30434-5.
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55, 2000607, doi:10.1183/13993003.00607-2020.
- Chen, J.; Wu, H.; Yu, Y.; Tang, N. Pulmonary alveolar regeneration in adult COVID-19 patients. Cell Res. 2020, 30, 708–710, doi:10.1038/s41422-020-0369-7.
- Qian, Z.; Travanty, E.A.; Oko, L.; Edeen, K.; Berglund, A.; Wang, J.; Ito, Y.; Holmes, K.V.; Mason, R.J. Innate Immune Re-sponse of Human Alveolar Type II Cells Infected with Severe Acute Respiratory Syndrome–Coronavirus. Am. J. Respir. Cell Mol. Biol. 2013, 48, 742–748, doi:10.1165/rcmb.2012-0339oc.
- Mason, R.J. Biology of alveolar type II cells. Respirology 2006, 11, S12–S15, doi:10.1111/j.1440-1843.2006.00800.x.
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal popula-tions contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483, doi:10.1073/pnas.1117988108.
- Rock, J.R.; Hogan, B. Epithelial Progenitor Cells in Lung Development, Maintenance, Repair, and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 493–512, doi:10.1146/annurev-cellbio-100109-104040.
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 1–7, doi:10.1186/s41232-020-00146-3.
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293, doi:10.1038/nm.4192.
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138, doi:10.1136/jcp.54.2.132.
- Young, L.R.; Pasula, R.; Gulleman, P.M.; Deutsch, G.H.; McCormack, F.X. Susceptibility of Hermansky-Pudlak Mice to Bleo-mycin-Induced Type II Cell Apoptosis and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 67–74, doi:10.1165/rcmb.2006-0469OC.
- Yu, F.; Jia, R.; Tang, Y.; Liu, J.; Wei, B. SARS-CoV-2 infection and stem cells: Interaction and intervention. Stem Cell Res. 2020, 46, 101859, doi:10.1016/j.scr.2020.101859.
- Valyaeva, A.A.; Zharikova, A.A.; Kasianov, A.S.; Vassetzky, Y.S.; Sheval, E.V. Expression of SARS-CoV-2 entry factors in lung epithelial stem cells and its potential implications for COVID-19. Sci. Rep. 2020, 10, 17772, doi:10.1038/s41598-020-74598-5.
- Hong, K.U.; Reynolds, S.D.; Watkins, S.; Fuchs, E.; Stripp, B.R. Basal Cells Are a Multipotent Progenitor Capable of Renewing the Bronchial Epithelium. Am. J. Pathol. 2004, 164, 577–588, doi:10.1016/s0002-9440(10)63147-1.
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The Role of Scgb1a1+ Clara Cells in the Long-Term Maintenance and Repair of Lung Airway, but Not Alveolar, Epithelium. Cell Stem Cell 2009, 4, 525–534, doi:10.1016/j.stem.2009.04.002.
- Rawlins, E.L.; Clark, C.P.; Xue, Y.; Hogan, B.L. The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development 2009, 136, 3741–3745, doi:10.1242/dev.037317.
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identifica-tion of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell 2005, 121, 823–835, doi:10.1016/j.cell.2005.03.032.
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46.
- Chen, Y.; Chan, V.S.; Zheng, B.; Chan, K.Y.; Xu, X.; To, L.Y.; Huang, F.P.; Khoo, U.S.; Lin, C.L. A novel subset of putative stem/progenitor CD34+Oct-4+ cells is the major target for SARS coronavirus in human lung. J. Exp. Med. 2007, 204, 2529–2536, doi:10.1084/jem.20070462.
- Ling, T.Y.; Kuo, M.D.; Li, C.L.; Yu, A.L.; Huang, Y.H.; Wu, T.J.; Lin, Y.C.; Chen, S.H.; Yu, J. Identification of pulmonary Oct-4+ stem/progenitor cells and demonstration of their susceptibility to SARS coronavirus (SARS-CoV) infection in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 9530–9535, doi:10.1073/pnas.0510232103.
- Quantius, J.; Schmoldt, C.; Vazquez-Armendariz, A.I.; Becker, C.; El Agha, E.; Wilhelm, J.; Morty, R.E.; Vadász, I.; Mayer, K.; Gattenloehner, S.; et al. Influenza Virus Infects Epithelial Stem/Progenitor Cells of the Distal Lung: Impact on Fgfr2b-Driven Epithelial Repair. PLoS Pathog. 2016, 12, e1005544, doi:10.1371/journal.ppat.1005544.
- Hancock, A.S.; Stairiker, C.J.; Boesteanu, A.C.; Monzón-Casanova, E.; Lukasiak, S.; Mueller, Y.M.; Stubbs, A.P.; García-Sastre, A.; Turner, M.; Katsikis, P.D. Transcriptome Analysis of Infected and Bystander Type 2 Alveolar Epithelial Cells during In-fluenza A Virus Infection Reveals In Vivo Wnt Pathway Downregulation. J. Virol. 2018, 92, doi:10.1128/jvi.01325-18.
- Hillesheim, A.; Nordhoff, C.; Boergeling, Y.; Ludwig, S.; Wixler, V. β-catenin promotes the type I IFN synthesis and the IFN-dependent signaling response but is suppressed by influenza A virus-induced RIG-I/NF-κB signaling. Cell Commun. Sig-nal. 2014, 12, 29, doi:10.1186/1478-811X-12-29.
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.-C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M.; et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020, 146, 206–208.e2, doi:10.1016/j.jaci.2020.04.029.
- Yang, X.; Zhao, C.; Bamunuarachchi, G.; Wang, Y.; Liang, Y.; Huang, C.; Zhu, Z.; Xu, D.; Lin, K.; Senavirathna, L.K.; et al. miR-193b represses influenza A virus infection by inhibiting Wnt/β-catenin signalling. Cell. Microbiol. 2019, 21, e13001, doi:10.1111/cmi.13001.
- Lin, F.-C.; Young, H.A. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 369–376, doi:10.1016/j.cytogfr.2014.07.015.
- Pan, H.; Peto, R.; Henao-Restrepo, A. M.; Preziosi, M. P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M. M.; Hernández García, C.; Kieny, M. P.; Malekzadeh, R.; Murthy, S., Reddy, K. S.; et al. Repurposed antiviral drugs for COVID-19—Interim WHO SOLIDARITY trial results. WHO Solidarity trial consortium. medRxiv 2020, doi:10.1101/2020.10.15.20209817.

