 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ekaterina Potekhina | + 4389 word(s) | 4389 | 2021-01-11 09:20:50 | | | |
| 2 | Nicole Yin | + 5 word(s) | 4394 | 2021-01-21 04:08:37 | | |
Video Upload Options
Genetically-encoded fluorescent sensors have been actively developed over the last few decades and used in live imaging and drug screening. Real-time monitoring of drug action in a specific cellular compartment, organ, or tissue type; the ability to screen at the single-cell resolution; and the elimination of false-positive results caused by low drug bioavailability that is not detected by in vitro testing methods are a few of the obvious benefits of using genetically-encoded fluorescent sensors in drug screening. In combination with high-throughput screening (HTS), some genetically encoded fluorescent sensors may provide high reproducibility and robustness to assays. Here, we provide an overview of attempts at using genetically encoded fluorescent sensors in HTS of anticancer compounds.
1. Introduction
Chemotherapy has developed greatly over the last few decades. However, the existing drugs often lack specificity, causing significant side effects, and the emergence of acquired drug resistance decreases drug efficiency at some point during therapy. Given these considerations, it is especially important to examine the efficacy, specificity, and pharmacodynamics of new drugs in live systems in a high-throughput real-time fashion. Genetically encoded fluorescent sensors meet these requirements, at least for some targets of anti-cancer therapy.
2. Kinase Inhibitors Screening
Protein kinase-dependent signaling plays a crucial role in the regulation of metabolism, cell cycle, differentiation, and death. The dysregulation of kinase activity is a significant factor in many pathological conditions, including oncological transformation, tumor growth, and metastasis. Therefore, protein kinases are considered a promising target for antitumor drugs. Specific kinase inhibitors are used for mono- or combinational antitumor therapy[1][2][3], but new drugs are required due to the frequent emergence of acquired drug resistance. Moreover, many kinase inhibitors compete with ATP for the binding pocket of the kinase. Since the structure of this pocket is very conservative among protein kinases, competitive inhibitors often lack specificity[4]. Different methods are used to screen the activity of kinases. These include the incorporation of radioactive phosphate containing 32P isotope, the use of phosphorylation-dependent antibodies, the use of non-genetic fluorescent peptide biosensors, and the use of genetically encoded biosensors based on fluorescent proteins[5]. The latter allow real-time monitoring of kinase signaling in cell cultures and tissues in a compartment-specific fashion[5][6][7]. Some genetically encoded fluorescent biosensors can be used not only for single-cell microscopy but also for high-content screening of novel inhibitor libraries.
In different forms of cancer, the tyrosine kinase Src is overexpressed or improperly regulated. This superfluous Src activity is involved in proliferation, adhesion, and invasive behavior of tumor cells[8]. A FRET-based Src indicator was designed and tested in vitro and in HeLa cells. This sensor consists of an Src substrate peptide from the p130cas molecule and a phosphotyrosine-binding SH2 domain from the c-Src molecule sandwiched between ECFP and EYFP. When the sensor is dephosphorylated, it remains in the “closed” conformation and the fluorescent proteins are juxtaposed, allowing energy transfer. After phosphorylation, the new linker conformation separates ECFP and EYFP, increasing the cyan-to-yellow emission ratio. The dynamic range of FRET with this sensor is 43%[9]. It was adapted for evaluating drug efficacy and delivery in the physiological tumor environment using multiphoton excitation and FLIM-FRET microscopy[10]. This approach is less sensitive to the loss of donor emission intensity caused by scattering in tissues[7] and accelerates data acquisition. Under these conditions, the Src indicator was able to mirror the spatial regulation of Src and the pharmacodynamics, delivery, and clearance of the tyrosine kinase inhibitor dasatinib in 3D tumor cultures and intravital tumor xenografts. For instance, in pancreatic ductal adenocarcinoma culture, invasive cell populations demonstrate higher Src activity than the upper surface of the culture. The same pattern was observed in vivo in xenografts, where Scr activity correlated with invasive regions and decreased in the center of the tumor. Dasatinib decreased Scr activity in invasive borders within 50 μm of the vasculature, but the regions in the center of the tumor, and/or more than 100 μm from the vasculature were poorly affected[10].
The Hippo pathway is involved in organogenesis, differentiation, and regeneration. In this pathway, the Ser/Thr LATS1/2 kinase phosphorylates several effector proteins including the growth-promoting transcriptional co-activator YAP. When phosphorylated on Ser127, YAP binds to cytosolic protein 14-3-3, which prevents YAP from transporting into the nucleus. The inactivation of LATS1/2 or upstream kinases and the amplification of YAP increases cell proliferation and decreases apoptosis and differentiation[11]. The biosensor for LATS (LATS-BS) activity is based on bioluminescence. The minimal YAP fragment capable of interacting with 14-3-3 in a phosphorylation-dependent manner is fused to the N-terminal luciferase fragment, while 14-3-3 is fused to the C-terminal luciferase fragment. When active LATS1/2 phosphorylates the YAP fragment, it binds to the 14-3-3 chimera, and active luciferase is assembled. LATS-BS was successfully used for a small-scale kinase inhibitor screen to identify the upstream regulators of the Hippo pathway. VEGFR, MEK, GSK-3, PKB/Akt, EGF receptor, and CDK 4 inhibitors (SU 4312, PD 98059, BIO, API-2, Genistein, and Ryuvidine, respectively) were shown to activate LATS-BS, while TrkA, SYK, ATR/ATM, CHK1, SGK, and broad-specific inhibitors (Ro 08-2750, ER 27319, CGK 733, PD 407824, GSK650394, and Ro 31-8220 respectively) reduced the LATS-BS signal. Some of these regulators had been previously described. However, the role of VEGFR was established de novo (Figure 1)[12].
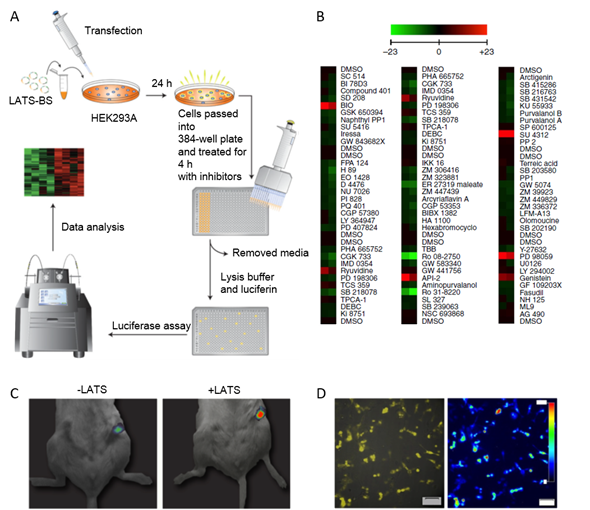
Figure 1. LATS-BS application to several experimental models. (A) Experimental design for high-throughput kinase inhibitor screen. Cells were transfected with LATS-BS, treated with a kinase inhibitor library, and luciferase assay was performed on cell lysates. (B) Heat map summarizing the results of the kinase inhibitor screen. Drugs that activate LATS-BS are shown in red, drugs that inhibit LATS-BS are shown in green. (C) LATS-BS was tested for in vivo imaging. HEK293 cells were transfected with LATS-BS only (-LATS) or LATS-BS and LATS kinase, injected into the mammary fat pad of immunocompromised mice, and bioluminescence was measured. (D) LATS-BS was used for bioluminescent imaging of LATS activity in live cells (MDA-MB231 cell line). The figures were taken from Azad et al. with minor changes[12].
The MAPK signaling network affects a wide range of cellular processes, including proliferation, metabolism, and apoptosis. In the MAPK/ERK cascade ERK functions as the final step, reflecting the activity of upstream elements. ERK substrates and final cellular response depend on the type of cell, spatiotemporal regulation, and activity of other signaling pathways. There are several biosensors for ERK activity. The prototype sensor ECAR, consists of mRFP1, a proline-directed WW phospho-binding domain, a peptide from Cdc25C containing the consensus MAPK target sequence, ERK docking peptide, and EGFP[13]. ERK shares the consensus substrate sequence with other MAP kinases[14]. However, the ERK docking motif provides binding and phosphorylation specificity. When phosphorylated, the sensor is in the “closed” conformation, and FRET occurs. Moreover, a CFP-YFP version containing Cerulean and Venus was produced. Both FRET pairs allow 2-photon fluorescence lifetime imaging. The CFP-YFP version allows FRET ratiometric measurement as well, providing almost the same signal-to-noise ratio[13]. After the application of an optimized EEVEE linker backbone, the ratiometric gain of the new sensor (EKAREV) increased fourfold and amounted to 40%[15]. Moreover, EKAREV was transformed into a hybrid FRET-BRET (hyBRET) sensor by fusing a bright Renilla luciferase mutant, RLuc8, to the C terminus of CFP (Turquoise2-GL). In the presence of substrate, the bioluminescence of RLuc8 excites CFP instead of external light sources. The BRET and FRET ratios correlate linearly, and the dynamic ranges in both measurement modes are almost equal. Due to the high signal-to-noise ratio, BRET sensors seem to be reliable instruments for automated drug screening. Indeed, hyBRET-ERK was able to quantify the dose-dependent response to MAPK pathway inhibitors in cancer cell lines cultured in microplates. For example, the IC50 for AZD6244, a MEK inhibitor, was determined using this method[16].
c-Jun N-terminal kinases (JNKs), also known as stress-activated protein kinases are another subfamily of MAP kinases. They are activated by environmental stress signals and cytokines and regulate apoptosis, inflammation, cell differentiation, and proliferation. JNKs are involved in cancer development, neurodegenerative diseases, insulin resistance, diabetes, and heart pathologies, thus JNK inhibitors might serve as drugs in these conditions[17][18][19][20]. JNK activity reporter (JNKAR1) consists of ECFP, forkhead associated domain 1 responsible for phosphoamino acid binding, a substrate sequence linked to the JDP2 docking domain, and Citrine. JNKAR1 is most probably sensitive to JNK1 and JNK 2 isoforms. After specific JNK activation in HeLa cells, the FRET ratio increases by up to 30%. It is noteworthy that the dephosphorylation of the reporter is slower than the dephosphorylation of ATF-2, an endogenous JNK substrate[21]. JNKAR1 has not been validated as an instrument for high-throughput screening. However, given that JNK is a promising drug target, and that the JNK pathway is likely to be a bistable system acting in an all-or-none fashion[21], JNKAR1 might be useful in drug screening assays. Moreover, JNKAR1 has been augmented with an optimized EEVEE linker, and the resulting JNKAR1EV construction demonstrated a dynamic FRET ratio range of about 100%[15].
The activity of the fusion protein Bcr-Abl produced by the Philadelphia chromosome causes chronic myelogenous leukemia (CML). Bcr-Abl is a constitutively active mutant of c-Abl tyrosine kinase. Inhibitors of tyrosine kinases such as imatinib mesylate and dasatinib are the main form of therapy for this condition. However, further mutations in Bcr-Abl make the tumor resistant to these drugs, and thus the development of novel inhibitors is required[22]. Two sensors detecting Bcr-Abl activity in vivo are available. The FRET-based sensor Picchu consists of C-terminally truncated adaptor protein Crk II flanked by CFP and YFP. Crk II Tyr221 is phosphorylated by Abl, EGFR[23], and Bcr-Abl[24] and binds to the SH2 domain, bringing the N-terminal YFP and C-terminal CFP together. The “closed” conformation allows resonance energy transfer from CFP to YFP [25]. The Picchu FRET ratio was demonstrated to reflect the activity of Bcr-Abl and the inhibitory effect and binding kinetics of imatinib and dasatinib. In cells expressing mutant Bcr-Abl forms, the FRET ratio was insensitive to inhibitors. The maximal change in FRET ratio was about 15–20%. However, this was sufficient to distinguish cells with active and inhibited Bcr-Abl by flow cytometry. According to the authors, Picchu can be used for automatic monitoring of drug resistance in mixed population clinical samples as well as for screening of potential next-generation Bcr-Abl inhibitors[24]. It is noteworthy that c-Abl phosphorylates Picchu with the same efficiency as Bcr-Abl[25].
Another sensor for Bcr-Abl activity is called Pickles. Its design exploits the same principles as Picchu. However, the sensitive domain is derived from CrkL, the most characteristic substrate of Bcr-Abl. The FRET pair is m1Venus and circularly permuted ECFP. In the presence of Bcr-Abl, FRET efficiency increases by 80%. Like Picchu, Pickles is phosphorylated not only by Bcr-Abl but also by c-Abl. However, the efficiency of these kinases for Pickles is different. Thus, Pickles is a more specific and sensitive indicator of Bcr-Abl activity. When expressed in cells, it was able to detect the effect, the dose-dependency, and the inhibition rate of Bcr-Abl inhibitors, such as imatinib, nilotinib, and dasatinib. Moreover, this sensor allowed evaluation of the drug response in primary human CML cells[25].
Receptors of growth factors, cytokines, and hormones (RTKs) are a large group of tyrosine kinases. Pathological activity of RTKs is associated with cancer emergence and progression, and the inhibitors of RTKs are widely used as therapeutic agents[26]. The fluorescent reporter of EGFR (epidermal growth factor receptor) activity named EGFRB is based on the specific mechanism of activation of RTKs. When bound to the ligand, RTKs form dimers where the monomers phosphorylate each other, and active receptors are internalized in endosomes. The sensor utilizes receptor clustering and endocytosis. It consists of GFP fused to two SH2 domains from Grb2 adapter protein, which are considered to have high affinity to phosphorylated EGFR. When the EGFRs are inactive, GFP fluorescence is homogeneously distributed throughout the cytosol. After EGFR activation and endocytosis, the sensor binds to the receptors, and fluorescent granules are observed. When expressed in the A549 cell line (human alveolar basal epithelial adenocarcinoma), the sensor demonstrated no response to RTK ligands except for EGF[27]. A549-EGFRB cells were validated as a system for high-content screening of EGFR modulators. After drug administration, the number of fluorescent granules in the wells of a microplate was counted. To measure cell number and cytotoxicity, nuclei stained by DRAQ5 were quantified. The EGFRB system demonstrated a high signal-to-noise ratio (21:1) and reproducibility. Using this system, a library of 6912 Food and Drug Administration-approved and known bioactive compounds was screened, and 12 of 13 reported EGFR kinase inhibitors from this library were picked as positive. Moreover, the Hsp90 inhibitor 17-DMAG was shown to decrease EGFR activity, most probably because Hsp90 is necessary for EGFR maturation. In the same screen, confirmed EGFR activators were shown to increase the number of granules. What is more, A549-EGFRB cells were successfully used for a dose-response test of EGFR modulators picked from the previous screen. According to the authors, EGFRB assay could be a potent instrument in EGFR modulator discovery[28].
3. Transcription Factors Regulators Screening
P53 takes part in the regulation of crucial cellular processes, including metabolism, cell cycle, stress response, and apoptosis. Decreased p53 activity due to mutations, misregulation, or enhanced decay is associated with tumorigenesis and cancer progression. Furthermore, p53 is ubiquitinylated and targeted to degradation by hDM2 protein, which is overexpressed in some types of cancer[29]. In order to find novel p53-hDM2 interaction inhibitors, a system for automated high-content screening for protein-protein interaction disruptor (PPID) was established. First, p53 was fused to GFP and augmented by an NLS localizing the construction in the nucleolus. hDM2 carried both NLS and NES and was fused to RFP. Naturally, both proteins colocalized in the nucleolus. In the presence of the known p53-hDM2 interaction inhibitor Nutlin-3, hDM2 was exported to the cytoplasm. A library of 220,000 small-molecule compounds was screened, and the assay demonstrated high reproducibility. However, a thorough analysis of fluorescence artifacts and cytotoxic effects was required to omit false-positive hits. Finally, three compounds related to methylbenzonaphthyridin-5-amine were confirmed to increase p53 protein level and apoptosis and to cause cell cycle arrest and growth inhibition in a p53-dependent fashion[30].
The gene c-Myc plays key roles in cell cycle regulation, growth, differentiation, and apoptosis. In the vast majority of cancers, this protein is abnormally abundant, stable, and active, which makes it a potential anti-cancer drug target. Mitogens and other stimulators activate c-Myc via Ser62 phosphorylation by ERK kinase. After stimulator removal, Ser62 recruits glycogen synthase kinase-3β (GSK-3β), which phosphorylates the Thr58 residue and targets c-Myc for proteasomal degradation[31]. c-Myc activation sensor consists of the c-Myc activation motif fused to the N-terminal domain of Renilla luciferase and GSK-3β phosphoamino acid binding domain fused to the C-terminal domain of Renilla luciferase. When c-Myc activation motif is phosphorylated, it binds to GSK-3β fragment, and the split luciferase fragments form an active enzyme. The sensor was tested in murine xenografts where it was able to reflect drug impact on c-Myc phosphorylation[32]. The sensor also proved to be applicable to high-throughput screening of c-Myc inhibitors. A library of 5000 compounds was tested on SKBR3 cells (breast cancer cell line with c-Myc overexpression), and about 1% of compounds were identified as clean positive hits, including known c-Myc pathway inhibitors, and nitazoxanide, a widely used antiprotozoan drug with few side effects. It should be mentioned that cell proliferation and luciferase activity inhibitors were also identified as positive hits. After further validation in c-Myc-associated cancer cell lines and xenografts, the c-Myc inhibiting and antineoplastic effects of nitazoxanide were confirmed[33].
4. Cell Death Signaling Inducers Screening
Another approach in anti-cancer drug discovery is the search for inducers of certain types of cell death, like apoptosis, mitotic catastrophe, or immunogenic cell death, irrespective of drug target. Some of these conditions can be detected using immunochemistry or chemical labeling. However, these methods are time- and labor-intensive and usually require fixation and permeabilization of cells. Thus, they are not well suited for high-throughput and in vivo assays. However, these phenomena can be observed in automated systems using genetically encoded fluorescent reporters.
For example, a system of two fluorescent reporters of mitotic catastrophe has been developed. Histone H2B-GFP chimera stably expressed in HCT 116 cells (human colon carcinoma) provides chromosome tracking, while Discosoma striata red fluorescent protein fused to centrin (DsRed-Centrin) visualizes the centrosomes, allowing polyploidy and multipolar divisions to be monitored. After plotting mean nuclear density against nuclear heterogeneity of H2B-GFP fluorescence, cells arrested in metaphase were observed as a distinct population, while other cells, including apoptotic ones, were distributed all over the plot. The assay was able to reflect mitotic catastrophe induction by several mitotic blockers, distinguish different cell fates after treatment, and show different susceptibility to the treatment in wild-type and p53−/− HCT cells. It is suitable for the microplate format and can be used for high-throughput detection of mitotic catastrophe[34].
The activation of effector caspase-3 is one of the most critical steps of apoptosis[35]. A FRET-based reporter of caspase-3 activity has been created. It consists of CFP, YFP, and a linker sequence containing the caspase-3 recognition site. After caspase-3 activation caused by UV radiation, toxic compounds, and other apoptotic stimuli, the sensor protein is cleaved, and the FRET ratio decreases. FRET ratio does not decrease in necrotic cell death. When expressed in HeLa-C3 cells grown in microplates, the sensor was able to dose-dependently reflect the pro-apoptotic effect for several compounds of known biological activity, such as vincristine, paclitaxel, and hydroxyurea, as well as for some novel plant-derived substances[36]. It should be mentioned, however, that YFP spectral properties are highly dependent on H+ and Cl− ion concentrations, and the latter may dramatically change during apoptosis. Venus fluorescent protein is more stable and can be used in such FRET-based sensors instead of YFP[37]. Moreover, the cleavage site DEVD, used in[36], can be recognized by other caspase-3-like proteases (DEVDases), for example, caspase-7[38].
Another approach for detection of caspase-3-like protease activity is using switch-on fluorescence-based caspase-3-like protease activity indicator (SFCAI). This sensor is based on a circular permutant of Venus protein, whose native N- and C-termini are connected by DEVDase recognition sequence. The artificial N- and C-termini are fused to Npu DnaE intein from Nostoc punctiforme, which catalyzes the trans-ligation of the termini, forming a cyclic protein. Cleavage by DEVDases alters the protein conformation and enables fluorescent activity. The sensor was tested in several cell cultures, and it was able to specifically reflect apoptotic DEVDases activation by cisplatin in a real-time and dose-dependent fashion. It is noteworthy that MCF-7 cells (human breast cancer) turned out to be the most convenient for fluorescent microscopy because they did not detach from the culture platform. The sensor also detected apoptosis in 3D cell cultures in soft agar. A group of relative sensors was developed based on different fluorescent proteins: superfolder GFP, Cerulean, and mCherry. According to the authors, SFCAIs could be used in high-throughput screening assays for pro-apoptotic agents due to their high sensitivity and robust signal[38].
Some apoptotic cells undergo membrane rupture after caspase activation entering secondary necrosis[39], so caspase activity reporters can leak from the cytoplasm, causing non-specific shifts in sensor readout. To distinguish between apoptosis and primary and secondary necrosis, one can use an additional fluorescent protein localized in membrane organoids, which stay intact during necrosis. For example, mitochondrion-targeted red fluorescent protein can be used together with a cleavable FRET-based caspase-3/7 activity reporter containing ECFP and EYFP. In apoptotic cells, the FRET ratio decreases, and in necrotic cells the FRET signal is lost, while RFP fluorescence remains constant. For cells stably expressing both reporters, high-throughput adaptable protocols for live-cell imaging and flow cytometry analysis are available[40].
Immunogenic cell death (ICD) inducers are another type of anti-cancer drugs. They stimulate cancer cells to emit signals that attract and activate immune cells. During premortem stress several processes occur, for example exposure of calreticulin on the surface of the cell, release of ATP, and the exodus of high-mobility group box 1 (HMGB1) protein from the nucleus to the cytoplasm[41]. LC3 protein migrates to autophagy-specific granules[42]. Using U2OS cells (human osteosarcoma) expressing calreticulin fused to RFP and HMGB1 or LC3 fused to GFP, a library of more than 500 compounds was screened. Cell nuclei were stained with DAPI, and nuclear pyknosis was also detected. This screening demonstrated that some tyrosine kinase inhibitors can induce ICD, which was further confirmed in cell cultures and murine xenografts[43]. HMGB1 exodus can also be observed via the “retention using selective hooks” (RUSH) system. In this system, U2OS cells express streptavidin fused to the NLS3 sequence and HMGB1 fused to streptavidin binding protein (SBP) and GFP. In the absence of biotin, the HMGB1-SBP-GFP chimera is bound to the streptavidin-NLS3 construction and localized in the nucleus. However, after ICD inducers and biotin treatment, GFP fluorescence is also observed in the cytosol. The assay is performed on fixed cells. It has been validated for high-throughput screening for ICD inducers. The RUSH system allows comparison of cell images with and without biotin treatment, thus filtering out false-positive results detected due to drug fluorescence and other unspecific factors[44]. This approach can also be applied to the search for protein secretion inhibitors[45].
The LC3-GFP reporter can be used in search of caloric restriction mimetics (CRM) as an autophagy reporter in tandem with protein acetylation monitoring. CRM can act as cardio- and hepatoprotectors and stimulate the immune response against tumor-associated antigens. LC3-GFP was used to identify CRM in a library of 200 compounds, and positive hits were tested for cytotoxicity and protein acetylation state. The screening revealed a CRM effect of 3,4-dimethoxychalcone, which was further confirmed to improve the T-cell dependent effect of the chemotherapeutic drug mitoxantrone[46].
5. Energy Metabolism Modulators Screening
Another promising area to target for anti-cancer anti-proliferative drugs is ATP production. Most cancer cells are characterized by metabolic changes, the Warburg effect is manifested as the predominance of glycolysis over oxidative phosphorylation[47]. The identification of small molecular inhibitors of glycolytic ATP production may be leveraged in the creation of some anti-cancer drugs. Imamura et al. developed ATeams FRET sensors for intracellular ATP level detection based on the epsilon subunit of Bacillus subtilis FOF1-ATP synthase fused to cyan fluorescent protein (mseCFP) at the N-terminus and yellow fluorescent protein (cp173-mVenus) at the C-terminus[48]. The physiological role of the ε-subunit is thought to be regulation of FOF1-ATP synthase activity depending on the intracellular ATP level. The subunit binds to ATP but does not hydrolyze it. In the ATP-bound form, the subunit retracts to draw the two fluorescent proteins close to each other, increasing FRET efficiency. ATeam version AT1.03 demonstrated a FRET signal dynamic range of about 2.5, and the detectable ATP concentration range was 1–8 mM. In 2018, Zhao et al. designed a three-step assay protocol in 96-well fluorescent plate reader format based on long-term transfectant K562 cells expressing ATeam FRET sensor version AT1.03, which allowed identification of compounds inhibiting glycolytic or OXPHOS-dependent ATP production, or both[49]. It was demonstrated that using 105 cells/well in 96-well fluorescent plate reader format made the variability of sensor FRET ratio between wells insignificant and the detected signal reproducible. The first read allowed detection of drugs that inhibit glucose-independent oxidative phosphorylation-dependent ATP production because measurements were made in the absence of glucose. The authors demonstrated that in those conditions K562 cells had produced ATP by OXPHOS using stored substrates for up to 3 h or until an inhibitor had been added. The second read was performed after 20 min incubation with 5 mM azide to block respiratory complex IV and provided an opportunity to select compounds that affect FRET ratios at the 1st read via fluorescence artifacts. Subsequent incubation with 20 mM D-glucose for 20 min preceded the third read. In this read, glycolysis inhibitors preventing ATP synthesis were detected. Unfortunately, the described method was not able to identify compounds that inhibit glycolytic or OXPHOS-dependent ATP production with a delayed mechanism of action. However, this FRET screening made it possible to test 813 compounds in the NCI’s (National Cancer Institute) Mechanistic Set III. ATP level influence was confirmed by luciferase ATP assay. The assay identified 14 compounds inhibiting oxidative phosphorylation-dependent ATP production by >75% and 13 compounds inhibiting glycolysis by >80%. Three of the detected inhibitors of oxidative phosphorylation, nonactin (NSC 52141), usnic acid (NSC 5890), and peliomycin (NSC 76455), were described previously. Other inhibitors were identified for the first time. ATeam FRET screening may be a basis for selecting inhibitors of ATP production with the ability to differentiate the path of ATP synthesis inhibition, i.e., glycolysis or OXPHOS. Significantly, Zhao et al. showed that the identified inhibitors of oxidative phosphorylation were not more antiproliferative than the NCI’s compound library as a whole, whereas glycolysis inhibitors were significantly more effective.
Due to the Warburg effect, many cancer cells show significantly lower NAD+/NADH ratios than non-cancer cells. The genetically encoded SoNar sensor was constructed by insertion of cpYFP between two subunits of NAD(H) binding protein Rex from Thermus aquaticus (T-Rex). NAD+ saturation of SoNar increases fluorescence exited at 485 nm, while NADH binding increases fluorescence exited at 420 nm. SoNar can specifically bind NAD+ or NADH (Kd ≈ 5.0 μM and ≈ 0.2 μM, respectively, at pH 7.4). It is a pH-stable, effective ratiometric sensor for NAD+, NADH, and their ratio. SoNar displays an 8-fold dynamic range in living cells. Zhao et al. presented SoNar as an excellent choice for HTS of the NAD+/NADH ratio in living cancer cells with drug screening opportunities[50]. More than 5500 unique compounds from the commercially available small-molecule libraries that might affect cellular metabolism were screened in the microplate format in H1299 cells using the SoNar sensor. Seventy-eight compounds were identified that significantly decreased the NAD+/NADH ratio in H1299 cells, and most of them did not exhibit obvious H1299 cell toxicity. Just 9 of 78 compounds were toxic. Compounds that significantly increased the NAD+/NADH ratio in H1299 cells exhibited H1299 cell toxicity (8 of 12 identified), among which were β-lapachone, shikonin, and fascaplysin, potent widely studied anti-tumor agents. The Akt inhibitor, also known as KP372-1, was identified as the compound with the most pronounced influence, increasing NAD+/NADH ratio, and with high cytotoxicity at 100 nM concentration for cancer cells of different origin, and with low toxicity toward various primary cells. KP372-1 was tested in H1299 xenografts expressing SoNar in mice, and the suppression of tumor growth was confirmed. Moreover, KP372-1 was shown to be cytotoxic to cancer cells independently from Akt inhibition. KP372-1 was proven as a potent NQO1-mediated redox cycling agent that causes severe oxidative stress and induces cancer cell apoptosis. The pronounced antitumor effect, bioavailability, and pharmacokinetics make KP372-1 a promising candidate for antitumor drugs.
References
- Amit Arora; Eric M. Scholar; Role of Tyrosine Kinase Inhibitors in Cancer Therapy. Journal of Pharmacology and Experimental Therapeutics 2005, 315, 971-979, 10.1124/jpet.105.084145.
- Yekaterina Y. Zaytseva; Joseph D. Valentino; Pat Gulhati; B. Mark Evers; mTOR inhibitors in cancer therapy. Cancer Letters 2012, 319, 1-7, 10.1016/j.canlet.2012.01.005.
- Christopher J. Caunt; Matthew J. Sale; Paul D. Smith; Simon J. Cook; MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nature Reviews Cancer 2015, 15, 577-592, 10.1038/nrc4000.
- Camille Prével; Morgan Pellerano; Thi Nhu Ngoc Van; May Catherine Morris; Fluorescent biosensors for high throughput screening of protein kinase inhibitors. Biotechnology Journal 2013, 9, 253-265, 10.1002/biot.201300196.
- Thi Nhu Ngoc Van; May C. Morris; Fluorescent Sensors of Protein Kinases. Progress in Molecular Biology and Translational Science 2013, 113, 217-274, 10.1016/b978-0-12-386932-6.00006-5.
- May Catherine Morris; Fluorescent biosensors — Probing protein kinase function in cancer and drug discovery. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2013, 1834, 1387-1395, 10.1016/j.bbapap.2013.01.025.
- Wei Lin; Sohum Mehta; Jin Zhang; Genetically encoded fluorescent biosensors illuminate kinase signaling in cancer. Journal of Biological Chemistry 2019, 294, 14814-14822, 10.1074/jbc.rev119.006177.
- Rosalyn B Irby; Timothy J Yeatman; Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636-5642, 10.1038/sj.onc.1203912.
- Yingxiao Wang; Elliot L. Botvinick; Yihua Zhao; Michael W. Berns; Shunichi Usami; Roger Y. Tsien; Shu Chien; Visualizing the mechanical activation of Src. Nature 2005, 434, 1040-1045, 10.1038/nature03469.
- Max Nobis; Ewan J. McGhee; Jennifer P. Morton; Juliane P. Schwarz; Saadia A. Karim; Jean Quinn; Mike Edward; Andrew D. Campbell; Lynn C. McGarry; T.R. Jeffry Evans; et al.Valerie G. BruntonMargaret C. FrameNeil O. CarragherYingxiao WangOwen J. SansomPaul TimpsonKurt I. Anderson Intravital FLIM-FRET Imaging Reveals Dasatinib-Induced Spatial Control of Src in Pancreatic Cancer. Cancer Research 2013, 73, 4674-4686, 10.1158/0008-5472.can-12-4545.
- Steven W. Plouffe; Audrey W. Hong; Kun-Liang Guan; Disease implications of the Hippo/YAP pathway. Trends in Molecular Medicine 2015, 21, 212-222, 10.1016/j.molmed.2015.01.003.
- T. Azad; H. J. Janse Van Rensburg; E. D. Lightbody; B. Neveu; Audrey Champagne; A. Ghaffari; V. R. Kay; Y. Hao; H. Shen; B. Yeung; et al.B. Anne CroyKun-Liang GuanF. PouliotJ. ZhangC. J. B. NicolXiaolong Yang A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nature Communications 2018, 9, 1-15, 10.1038/s41467-018-03278-w.
- Christopher D. Harvey; Anka G. Ehrhardt; Cristina Cellurale; Haining Zhong; Ryohei Yasuda; Roger J. Davis; Karel Svoboda; A genetically encoded fluorescent sensor of ERK activity. Proceedings of the National Academy of Sciences 2008, 105, 19264-19269, 10.1073/pnas.0804598105.
- Marie A. Bogoyevitch; Bostjan Kobe; Uses for JNK: the Many and Varied Substrates of the c-Jun N-Terminal Kinases. Microbiology and Molecular Biology Reviews 2006, 70, 1061-1095, 10.1128/mmbr.00025-06.
- Naoki Komatsu; Kazuhiro Aoki; Masashi Yamada; Hiroko Yukinaga; Yoshihisa Fujita; Yuji Kamioka; Michiyuki Matsuda; Development of an optimized backbone of FRET biosensors for kinases and GTPases. Molecular Biology of the Cell 2011, 22, 4647-4656, 10.1091/mbc.e11-01-0072.
- Naoki Komatsu; Kenta Terai; Ayako Imanishi; Yuji Kamioka; Kenta Sumiyama; Takashi Jin; Yasushi Okada; Takeharu Nagai; Michiyuki Matsuda; A platform of BRET-FRET hybrid biosensors for optogenetics, chemical screening, and in vivo imaging. Scientific Reports 2018, 8, 1-14, 10.1038/s41598-018-27174-x.
- Brydon Bennett; JNK: a new therapeutic target for diabetes. Current Opinion in Pharmacology 2003, 3, 420-425, 10.1016/s1471-4892(03)00068-7.
- Tiziana Borsello; Gianluigi Forloni; JNK signalling: a possible target to prevent neurodegeneration.. Current Pharmaceutical Design 2007, 13, 1875-1886, 10.2174/138161207780858384.
- Beth A. Rose; Thomas Force; Yibin Wang; Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiological Reviews 2010, 90, 1507-1546, 10.1152/physrev.00054.2009.
- Concetta Bubici; Salvatore Papa; JNK signalling in cancer: in need of new, smarter therapeutic targets. British Journal of Pharmacology 2013, 171, 24-37, 10.1111/bph.12432.
- Matthew Fosbrink; Nwe-Nwe Aye-Han; Raymond Cheong; Andre Levchenko; Jin Zhang; Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proceedings of the National Academy of Sciences 2010, 107, 5459-5464, 10.1073/pnas.0909671107.
- Federico Rossari; Filippo Minutolo; Enrico Orciuolo; Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. Journal of Hematology & Oncology 2018, 11, 1-14, 10.1186/s13045-018-0624-2.
- Kazuo Kurokawa; Naoki Mochizuki; Yusuke Ohba; Hideaki Mizuno; Atsushi Miyawaki; Michiyuki Matsuda; A Pair of Fluorescent Resonance Energy Transfer-based Probes for Tyrosine Phosphorylation of the CrkII Adaptor Proteinin Vivo. Journal of Biological Chemistry 2001, 276, 31305-31310, 10.1074/jbc.m104341200.
- Ahmet Tunceroglu; Michiyuki Matsuda; Raymond B. Birge; Real-time Fluorescent Resonance Energy Transfer Analysis to Monitor Drug Resistance in Chronic Myelogenous Leukemia. Molecular Cancer Therapeutics 2010, 9, 3065-3073, 10.1158/1535-7163.mct-10-0623.
- Tatsuaki Mizutani; Takeshi Kondo; Stephanie Darmanin; Masumi Tsuda; Kazuma Tanaka; Minoru Tobiume; Masahiro Asaka; Yusuke Ohba; A Novel FRET-Based Biosensor for the Measurement of BCR-ABL Activity and Its Response to Drugs in Living Cells. Clinical Cancer Research 2010, 16, 3964-3975, 10.1158/1078-0432.ccr-10-0548.
- Andreas Gschwind; Oliver M. Fischer; Axel Ullrich; The discovery of receptor tyrosine kinases: targets for cancer therapy. Nature Cancer 2004, 4, 361-370, 10.1038/nrc1360.
- Christophe Antczak; Alun Bermingham; Paul Calder; Dmitry Malkov; Keming Song; John Fetter; Hakim Djaballah; Domain-Based Biosensor Assay to Screen for Epidermal Growth Factor Receptor Modulators in Live Cells. ASSAY and Drug Development Technologies 2012, 10, 24-36, 10.1089/adt.2011.423.
- Christophe Antczak; Jeni P. Mahida; Bhavneet Bhinder; Paul A. Calder; Hakim Djaballah; A High-Content Biosensor-Based Screen Identifies Cell-Permeable Activators and Inhibitors of EGFR Function. Journal of Biomolecular Screening 2012, 17, 885-899, 10.1177/1087057112446174.
- Ute M. Moll; Oleksi Petrenko; The MDM2-p53 interaction.. Molecular Cancer Research 2003, 1, 1001-1008, PMID: 14707283.
- Drew D. Dudgeon; Sunita Shinde; Yun Hua; Tong Ying Shun; John S. Lazo; Christopher J. Strock; Kenneth A. Giuliano; D. Lansing Taylor; Patricia A. Johnston; Paul A. Johnston; et al. Implementation of a 220,000-Compound HCS Campaign to Identify Disruptors of the Interaction between p53 and hDM2 and Characterization of the Confirmed Hits. Journal of Biomolecular Screening 2010, 15, 766-782, 10.1177/1087057110375304.
- Elizabeth Yeh; Melissa Cunningham; Hugh Arnold; Dawn A D Chasse; Teresa Monteith; Giovanni Paolo Ivaldi; William C. Hahn; P. Todd Stukenberg; Shirish Shenolikar; Takafumi Uchida; et al.Christopher M. CounterJoseph R. NevinsAnthony R. MeansRosalie C Sears A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nature 2004, 6, 308-318, 10.1038/ncb1110.
- Hua Fan-Minogue; Zhongwei Cao; Ramasamy Paulmurugan; Carmel T. Chan; Tarik F. Massoud; Dean W. Felsher; Sanjiv S. Gambhir; Noninvasive molecular imaging of c-Myc activation in living mice. Proceedings of the National Academy of Sciences 2010, 107, 15892-15897, 10.1073/pnas.1007443107.
- Hua Fan-Minogue; Sandhya Bodapati; David Solow-Cordero; Alice Fan; Ramasamy Paulmurugan; Tarik F. Massoud; Dean W. Felsher; Sanjiv S. Gambhir; A c-Myc Activation Sensor-Based High-Throughput Drug Screening Identifies an Antineoplastic Effect of Nitazoxanide. Molecular Cancer Therapeutics 2013, 12, 1896-1905, 10.1158/1535-7163.mct-12-1243.
- Santiago Rello-Varona; O Kepp; Ilio Vitale; M Michaud; Laura Senovilla; Mohamed Jemaà; N Joza; L Galluzzi; M Castedo; Guido Kroemer; et al. An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death & Disease 2010, 1, e25-e25, 10.1038/cddis.2010.6.
- Alan G Porter; Reiner U Jänicke; Emerging roles of caspase-3 in apoptosis. Cell Death & Differentiation 1999, 6, 99-104, 10.1038/sj.cdd.4400476.
- Honglei Tian; Louisa Ip; Houwei Luo; Donald C. Chang; Kathy Q. Luo; A high throughput drug screen based on fluorescence resonance energy transfer (FRET) for anticancer activity of compounds from herbal medicine. British Journal of Pharmacology 2007, 150, 321-334, 10.1038/sj.bjp.0706988.
- Kiwamu Takemoto; Takeharu Nagai; Atsushi Miyawaki; Masayuki Miura; Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effects. The Journal of Cell Biology 2003, 160, 235-243, 10.1083/jcb.200207111.
- Jiao Zhang; Xin Wang; Wenjing Cui; Wenwen Wang; Huamei Zhang; Lu Liu; Zicheng Zhang; Zheng Li; Guoguang Ying; Ning Zhang; et al.Binghui Li Visualization of caspase-3-like activity in cells using a genetically encoded fluorescent biosensor activated by protein cleavage. Nature Communications 2013, 4, 2157, 10.1038/ncomms3157.
- Manuel T. Silva; Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Letters 2010, 584, 4491-4499, 10.1016/j.febslet.2010.10.046.
- Asha Lekshmi; Shankara Narayanan Varadarajan; Santhik Subhasingh Lupitha; Mydhily Nair; Aneesh Chandrasekharan; T.R. Santhoshkumar; A Real‐Time Image‐Based Approach to Distinguish and Discriminate Apoptosis from Necrosis. Current Protocols in Toxicology 2018, 75, 2.27.1-2.27.16, 10.1002/cptx.39.
- Guido Kroemer; Lorenzo Galluzzi; Oliver Kepp; Laurence Zitvogel; Immunogenic Cell Death in Cancer Therapy. Annual Review of Immunology 2013, 31, 51-72, 10.1146/annurev-immunol-032712-100008.
- Isei Tanida; Takashi Ueno; Eiki Kominami; LC3 and Autophagy. Toxicity Assessment 2008, 445, 77-88, 10.1007/978-1-59745-157-4_4.
- Peng Liu; Liwei Zhao; Jonathan Pol; Sarah Levesque; Adriana Petrazzuolo; Christina Pfirschke; Camilla Engblom; Steffen Rickelt; Takahiro Yamazaki; Kristina Iribarren; et al.Laura SenovillaLucillia BezuErika VacchelliValentina SicaAndréa MelisTiffany MartinLin XiaHeng YangQingqing LiJinfeng ChenSylvère DurandFanny AprahamianDeborah LefevreSophie BroutinAngelo PaciAmaury BongersVeronique Minard-ColinEric TartourLaurence ZitvogelLionel ApetohYuting MaMikael J. PittetOliver KeppGuido Kroemer Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nature Communications 2019, 10, 1486, 10.1038/s41467-019-09415-3.
- Liwei Zhao; Peng Liu; Oliver Kepp; Guido Kroemer; Methods for measuring HMGB1 release during immunogenic cell death. Methods in Enzymology 2019, 629, 177-193, 10.1016/bs.mie.2019.05.001.
- Liwei Zhao; Peng Liu; Gaelle Boncompain; Friedemann Loos; Sylvie Lachkar; Lucillia Bezu; Guo Chen; Heng Zhou; Franck Perez; Oliver Kepp; et al.Guido Kroemer Identification of pharmacological inhibitors of conventional protein secretion. Scientific Reports 2018, 8, 14966, 10.1038/s41598-018-33378-y.
- Guo Chen; Wei Xie; Jihoon Nah; Allan Sauvat; Peng Liu; Federico Pietrocola; Valentina Sica; Didac Carmona‐Gutierrez; Andreas Zimmermann; Tobias Pendl; et al.Jelena TadicMartina BergmannSebastian J. HoferLana DomuzSylvie LachkarMaria MarkakiNektarios TavernarakisJunichi SadoshimaFrank MadeoOliver KeppGuido Kroemer 3,4‐Dimethoxychalcone induces autophagy through activation of the transcription factors TFE 3 and TFEB. EMBO Molecular Medicine 2019, 11, e10469, 10.15252/emmm.201910469.
- Jie Zheng; Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncology Letters 2012, 4, 1151-1157, 10.3892/ol.2012.928.
- Hiromi Imamura; Kim P. Huynh Nhat; Hiroko Togawa; Kenta Saito; Ryota Iino; Yasuyuki Kato-Yamada; Takeharu Nagai; Hiroyuki Noji; Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proceedings of the National Academy of Sciences 2009, 106, 15651-15656, 10.1073/pnas.0904764106.
- Ziyan Zhao; Rahul Rajagopalan; Adam Zweifach; A Novel Multiple-Read Screen for Metabolically Active Compounds Based on a Genetically Encoded FRET Sensor for ATP. SLAS DISCOVERY: Advancing the Science of Drug Discovery 2018, 23, 907-918, 10.1177/2472555218780636.
- Yuzheng Zhao; Qingxun Hu; Feixiong Cheng; Ni Su; Aoxue Wang; Yejun Zou; Hanyang Hu; Xianjun Chen; Hai-Meng Zhou; Xinzhi Huang; et al.Kai YangQian ZhuXue WangJing YiLinyong ZhuXuhong QianLixin ChenYun TangJoseph LoscalzoYi Yang SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metabolism 2015, 21, 777-789, 10.1016/j.cmet.2015.04.009.


