 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agostinho Lemos | + 1555 word(s) | 1555 | 2020-04-15 12:27:23 | | | |
| 2 | Camila Xu | -49 word(s) | 1506 | 2020-11-01 09:44:30 | | |
Video Upload Options
The suitability of the 18F radioisotope in positron emission tomography (PET) demanded novel approaches for 18F-fluorination and 18F-fluoroalkylation. The difluoromethyl (CF2H) group has gained increasing attention in medicinal chemistry due to its lipophilic hydrogen-bond donor properties. In non-radioactive chemistry, difluoromethyl heteroaryl-sulfones has been extensively used in difluoromethylation of substrates bearing C=C, C≡C, and C≡N bonds by visible light photoredox catalysis. Herein, we highlight our recent work on the synthesis of [18F]difluoromethyl heteroaryl-sulfones with improved molar activities and their application in photoinduced C-H 18F-difluoromethylation of N-containing heteroarenes via a radical-mediated pathway.
1. Introduction
The fluorine-18 radioisotope has been regarded the "radionuclide of choice" for in vivo positron emission tomography (PET) imaging in living subjects since it provides a unique combination of nuclear and physical features over alternative short-lived positron emitters such as carbon-11, nitrogen-13, and oxygen-15[1][2][3][4]. Fluorine-18 has a high positron yield resulting from an almost exclusive β+ emission (97%) with this mode of decay producing the stable oxygen-18 isotope. The half-life of 109.8 min allows sufficient time for multistep synthetic labeling reactions and the transport of 18F-labeled radiotracers over considerable distances. Furthermore, the relatively low positron energy of fluorine-18 (0.635 MeV) is highly advantageous in the acquisition of high-resolution PET images[1][2][3][4]. The distinctive sensitivity of PET makes this technique suitable for the study of absorption, distribution, metabolism, and excretion (ADME) properties of radiopharmaceuticals and the evaluation of their pharmacodynamic profile. In addition, PET technology has proven highly valuable in the observation of biochemical and physiological changes that may take place before the anatomical alterations of a certain disease are detected[5][6][7][8]. The suitability of the 18F radioisotope in PET has encouraged radiochemists to invest much effort in the development of efficient 18F-fluorination and 18F-fluoroalkylation reactions[9][10][11].
Among the existing fluorinated motifs, the difluoromethyl (CF2H) group has received considerable attention in medicinal chemistry due to its lipophilic hydrogen-bond donor properties. The CF2H substitution may offer a viable alternative to conventional hydrogen-bond donors [e.g., hydroxy (OH) and thiol (SH) groups] in terms of lipophilicity, cell membrane permeability, and metabolic stability, thus modulating the pharmacological activity of pharmaceuticals and agrochemicals[12][13]. Thus, the synthesis of radiotracers with 18F-difluoromethyl groups has been recently explored due to the attractive characteristics of fluorine-18 and CF2H motifs in radiopharmaceutical chemistry.
2. Overview of the different strategies for the synthesis of [18F]aryl-CF2H derivatives
Despite the recent progresses in the preparation of CF2H-containing derivatives in organofluorine chemistry[14][15][16], methodologies for the 18F-labeling of CF2H groups are still relatively scarce and mainly relied on the radiosynthesis of [18F]aryl–CF2H derivatives via 18F-fluorination reactions (Figure 1). In 2013, Gouverneur and co-workers disclosed a silver(I)-mediated protocol enabling the 18F-fluorodecarboxylation of 2-fluoro-2-arylacetic acids with [18F]Selectfluor bis(triflate), an electrophilic 18F-fluorinating reagent[17] (Scheme 1A). The cyclotron-produced [18F]fluoride was employed for the first time to access [18F]aryl–CF2H derivatives by AgOTf-mediated 18F-fluorination of aryl-CHFCl precursors[18] (Scheme 1B). In 2016, Ritter developed an alternative approach for the construction of [18F]aryl–CF2H functionalities starting from aryl (pseudo)halides via activation of a benzoyl auxiliary followed by benzylic bromination and in situ halogen exchange with [18F]fluoride[19] (Scheme 1C). Later, Liang and co-workers described an alternative approach for the preparation of [18F]aryl–CF2H with improved molar activity[20]. This synthetic approach consisted in the initial nucleophilic 18F-fluorination of benzyl (pseudo)halides and subsequent oxidative benzylic C-H fluorination with Selectfluor under transition metal-free conditions (Scheme 1D). In 2019, Gouverneur reported the utilization of aryl boronic acids as substrates to afford [18F]aryl-CF2H through copper-mediated cross-coupling with ethyl bromofluoroacetate and manganese-mediated 18F-fluorodecarboxylation with [18F]tetraethylammonium fluoride ([18F]TEAF) and iodosobenzene (PhIO)[21] (Scheme 1E).
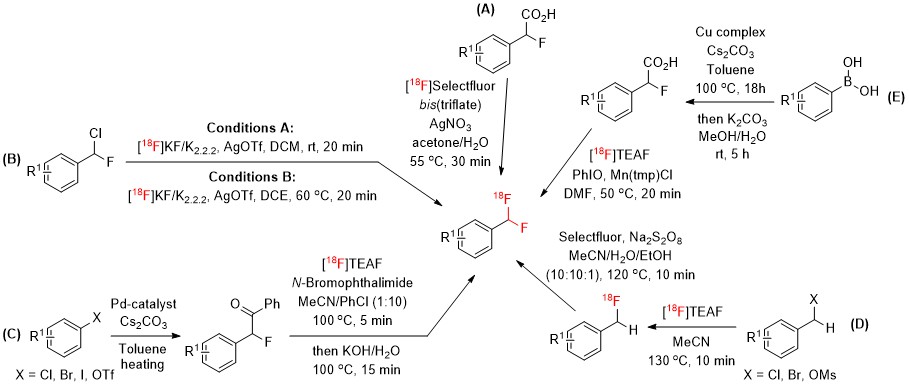
These methods allowed the radiosynthesis of [18F]aryl–CF2H derivatives with low-to-moderate molar activities (up to 22 GBq·μmol-1). The production of radiotracers with high molar activity is mandatory for PET imaging studies, especially for targetting low-density biomacromolecules. Obtaining a high molar activity still constitutes a major challenge in the preparation of [18F]CF2H–bearing compounds, due to the unwanted 18F–19F isotopic exchange reactions.
3. Radiosynthesis of [18F]difluoromethyl heteroaryl-sulfones for C-H 18F-difluoromethylation of heteroarenes by visible light photoredox catalysis
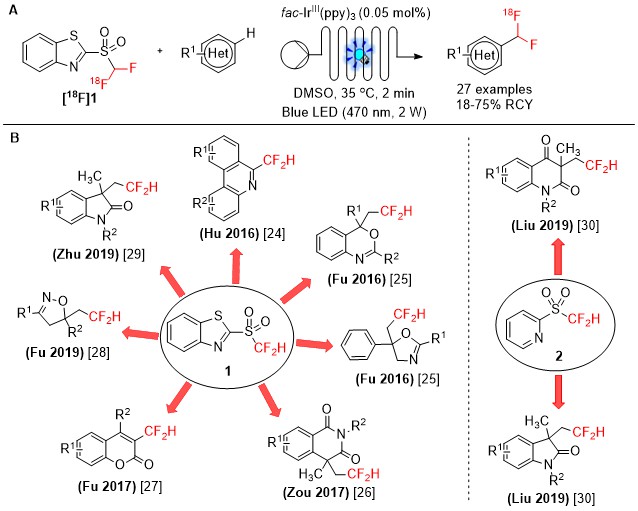
In 2019, our laboratories described an innovative and efficient protocol enabling the late-stage introduction of 18F-difluoromethyl moieties in N-containing heteroarenes with the [18F]difluoromethyl benzothiazolyl-sulfone ([18F]1) with improved molar activity [Am ([18F]1) = 54 ± 7 GBq·μmol-1][22][23] (Figure 1A). In non-radioactive chemistry, difluoromethyl heteroaryl-sulfones have been extensively employed in photoredox-catalyzed difluoromethylation of substrates bearing C=C, C≡C, and C≡N bonds because of the ability of these compounds to be reduced to CF2H radicals in the presence of appropriate photocatalysts in their photoexcited state. In 2016, Hu and Fu reported the use of difluoromethyl benzothiazolyl-sulfone (1) in the radical difluoromethylation of biphenyl isocyanides[24] and olefinic amides[25], respectively. The reagent 1 was also employed in the preparation of CF2H–substituted heterocycles of biological relevance, including isoquinolinediones[26], coumarins[27], isoxazolines[28], and oxindoles[29]. In 2019, Liu and co-workers developed a procedure for the difluoromethylation of N-arylacrylamides with the reagent difluoromethyl pyridyl-sulfone (2), under visible light photoredox conditions[30] (Figure 1B).
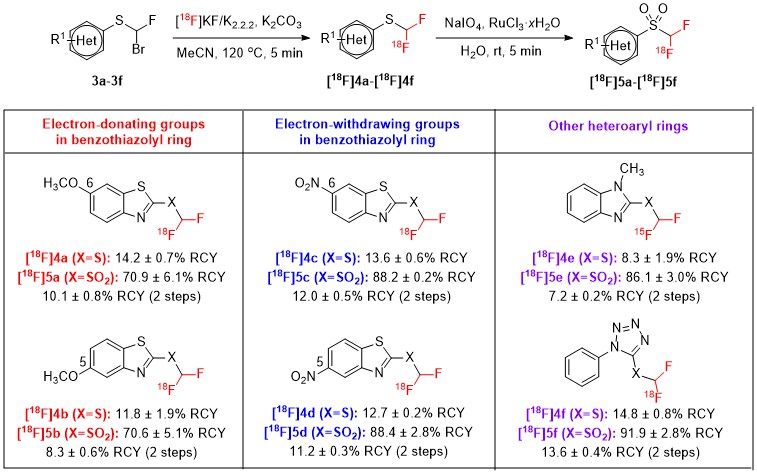
Taking advantage of the reported efficiency of the [18F]1 as 18F-difluoromethylating reagent[22][23], our laboratories planned to study the influence of certain molecular modifications in the structure of [18F]1 in the reactivity toward the photoredox C-H 18F-difluoromethylation of heteroarenes, under continuous-flow conditions[31]. The molecular modifications consisted in the single introduction of an electron-donating (EDG) or an electron-withdrawing group (EWG) either at position 5 or 6 of the benzothiazolyl ring and the modification of the original benzothiazolyl moiety to other heteroaryl rings (N-methyl-benzimidazolyl and N-phenyl-tetrazolyl rings). Six structurally-related [18F]difluoromethyl heteroaryl-sulfones ([18F]5a-[18F]5f) were synthesized on a GE FASTlabTM synthesizer via a two-step sequence (Scheme 2). The nucleophilic 18F-fluorination of bromofluoromethyl heteroaryl-sulfides (3a-3f) was carried out in the presence of potassium carbonate (K2CO3) and Kryptofix® 222 (K2.2.2), and afforded the cartridge-purified [18F]difluoromethyl heteroaryl-sulfides ([18F]4a-[18F]4f) in 8.3–14.8% RCYs [decay-corrected at the start-of-synthesis (SOS)]. After optimization of the oxidation step with sodium (meta)periodate (NaIO4) and ruthenium (III) chloride hydrate (RuCl3·xH2O), the sulfones [18F]5a-[18F]5f were afforded in 70.6–91.9% RCYs (decay-corrected at the SOS).
Based on the RCYs of the two-step radiosyntheses of the cartridge-purified [18F]5a–[18F]5f, the electron-rich benzothiazolyl derivative [18F]5a, the electron-poor benzothiazolyl derivative [18F]5c, and the N-phenyl tetrazolyl derivative [18F]5f were selected for the investigation of their reactivity toward 18F-difluoromethylation of N-containing heteroarenes. In order to circumvent any potential radioprotection issues, the radiosyntheses of the three 18F-labeled compounds were fully automated on a FASTlabTM synthesizer (GE Healthcare) in conjunction with an additional high performance liquid chromatography (HPLC) purification. The fully automated radiosyntheses of [18F]5a, [18F]5c, and [18F]5f (18F-labeling, oxidation, HPLC purification, and formulation) were performed in 73 min, 70 min, and 65 min, respectively. Starting from 125-150 GBq of [18F]fluoride, the [18F]5a, [18F]5c, and [18F]5f were isolated in 2.9 ± 0.1%, 5.7 ± 0.5%, and 8.0 ± 0.9% RCYs (decay-corrected at the SOS), respectively. Improved molar activities at the end of the synthesis (EOS) were achieved under these radiolabeling conditions [Am ([18F]5a) = 139 ± 17 GBq·μmol−1 > Am ([18F]5f) = 113 ± 17 GBq·μmol−1 > Am ([18F]5c) = 62 ± 12 GBq·μmol−1 > Am ([18F]1) = 54 ± 7 GBq·μmol−1] (Table 1).
Table 1. Radiochemical yields and molar activities of [18F]5a, [18F]5c, and [18F]5f.
| [18F]Difluoromethyl Heteroaryl-Sulfones | [18F]5a | [18F]5c | [18F]5f |
| Duration of the radiosynthesis (min) | 73 | 70 | 65 |
| RCY (%) | 2.9 ± 0.1 | 5.7 ± 0.5 | 8.0 ± 0.9 |
| Molar activity (GBq·μmol-1) | 139 ± 17 | 62 ± 12 | 113 ± 17 |
Subsequently, the efficiency of [18F]5a, [18F]5c, and [18F]5f as 18F-difluoromethylating reagents was evaluated using the antiherpetic drug acyclovir as a model substrate. The 18F-difluoromethylation reactions were carried out under continuous-flow conditions, in the presence of the photocatalyst fac-IrIII(ppy)3. Our results demonstrated that the introduction of molecular modifications in the structure of [18F]1 can modulate the reactivity of the resulting 18F-difluoromethylating reagents, influencing the amount of fac-IrIII(ppy)3 and the residence time needed to ensure a complete C-H 18F-difluoromethylation reaction. The photocatalytic 18F-difluoromethylation protocol with the reagents [18F]5a, [18F]5c, and [18F]5f was extended to other heteroarenes (Scheme 3; [18F]5a: 17-57% RCYs; [18F]5c: 13-66% RCYs; [18F]5f: 14-60% RCYs). Radical-trapping experiments demonstrated the likely involvement of radical species in the 18F-difluoromethylation process.
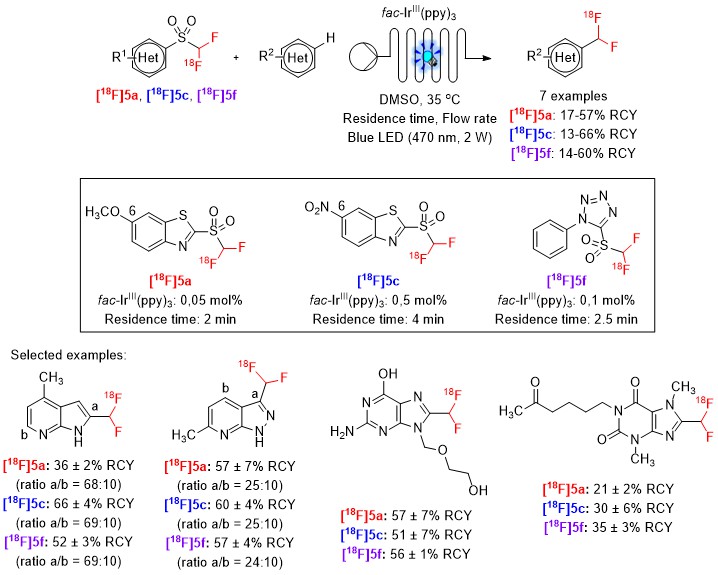
Scheme 3. Scope of [18F]heteroaryl-CF2H derivatives. All RCYs were determined based on the radio-TLC and radio-UPLC purities of the crude products.
References
- Didier Le Bars; Fluorine-18 and medical imaging: Radiopharmaceuticals for Positron Emission Tomography. Journal of Fluorine Chemistry 2006, 127, 1488-1493, 10.1016/j.jfluchem.2006.09.015.
- Einat Even-Sapir; Eyal Mishani; Gideon Flusser; Ur Metser; 18F-Fluoride Positron Emission Tomography and Positron Emission Tomography/Computed Tomography. Seminars in Nuclear Medicine 2007, 37, 462-469, 10.1053/j.semnuclmed.2007.07.002.
- Samuel D. Banister; Dirk Roeda; Frédéric Dollé; Michael Kassiou; Fluorine-18 Chemistry for PET: A Concise Introduction. Current Radiopharmaceuticals 2010, 3, 68-80, 10.2174/1874471011003020068.
- Cassis Varlow; Daniel Szames; Kenneth Dahl; Vadim Bernard-Gauthier; Neil Vasdev; Fluorine-18: An Untapped Resource in Inorganic Chemistry. Chemical Communications 2018, 54, 11835-11842, 10.1039/c8cc04751k.
- A. K. Shukla; Utham Kumar; Positron Emission Tomography: An Overview. Journal of Medical Physics 2006, 31, 13-21, 10.4103/0971-6203.25665.
- M. Lin; I. Ho Shon; P. Lin; Positron Emission Tomography: Current Status and Future Challenges. Internal Medicine Journal 2010, 40, 19-29, 10.1111/j.1445-5994.2009.02072.x.
- Juan José Vaquero; Paul Kinahan; Positron Emission Tomography: Current Challenges and Opportunities for Technological Advances in Clinical and Preclinical Imaging Systems. Annual Review of Biomedical Engineering 2015, 17, 385-414, 10.1146/annurev-bioeng-071114-040723.
- Tarec C. El-Galaly; Lars Christian Gormsen; Martin Hutchings; PET/CT for Staging; Past, Present, and Future. Seminars in Nuclear Medicine 2018, 48, 4-16, 10.1053/j.semnuclmed.2017.09.001.
- Sean Preshlock; Matthew Tredwell; Véronique Gouverneur; 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chemical Reviews 2016, 116, 719-766, 10.1021/acs.chemrev.5b00493.
- Dion Van Der Born; Anna Pees; Alex Poot; Romano V. A. Orru; Albert D. Windhorst; Danielle J. Vugts; Fluorine-18 labelled building blocks for PET tracer synthesis. Chemical Society Reviews 2017, 46, 4709-4773, 10.1039/C6CS00492J.
- Xiaoyun Deng; Jian Rong; Lu Wang; Neil Vasdev; Lei Zhang; Lee Josephson; Steven H. Liang; Chemistry for Positron Emission Tomography: Recent Advances in 11 C‐, 18 F‐, 13 N‐, and 15 O‐Labeling Reactions. Angewandte Chemie International Edition 2019, 58, 2580-2605, 10.1002/anie.201805501.
- Yossi Zafrani; Dina Yeffet; Gali Sod-Moriah; Anat Berliner; Dafna Amir; Daniele Marciano; Eytan Gershonov; Sigal Saphier; Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. Journal of Medicinal Chemistry 2017, 60, 797-804, 10.1021/acs.jmedchem.6b01691.
- Chanan D. Sessler; Martin Rahm; Sabine Becker; Jacob M. Goldberg; Fang Wang; Stephen J. Lippard; CF2H, a Hydrogen Bond Donor. Journal of the American Chemical Society 2017, 139, 9325-9332, 10.1021/jacs.7b04457.
- Jian Rong; Chuanfa Ni; Jinbo Hu; Metal-Catalyzed Direct Difluoromethylation Reactions. Asian Journal of Organic Chemistry 2017, 6, 139-152, 10.1002/ajoc.201600509.
- Damian E. Yerien; Sebastian Barata-Vallejo; Al Postigo; Difluoromethylation Reactions of Organic Compounds. Chemistry - A European Journal 2017, 23, 14676-14701, 10.1002/chem.201702311.
- Agostinho Lemos; Christian Lemaire; André Luxen; Progress in Difluoroalkylation of Organic Substrates by Visible Light Photoredox Catalysis. Advanced Synthesis & Catalysis 2019, 361, 1500-1537, 10.1002/adsc.201801121.
- Satoshi Mizuta; Ida S. R. Stenhagen; Miriam L. O'duill; Jamie Wolstenhulme; Anna K Kirjavainen; Sarita J. Forsback; Matthew Tredwell; Graham Sandford; Peter R. Moore; Mickael Huiban; et al.Sajinder K. LuthraJan PasschierOlof SolinVéronique Gouverneur Catalytic Decarboxylative Fluorination for the Synthesis of Tri- and Difluoromethyl Arenes. Organic Letters 2013, 15, 2648-2651, 10.1021/ol4009377.
- Véronique Gouverneur; Stefan Verhoog; Lukas Pfeifer; Tanatorn Khotavivattana; Samuel Calderwood; Thomas Collier; Katherine Wheelhouse; Matthew Tredwell; Silver-Mediated 18F-Labeling of Aryl-CF3 and Aryl-CHF2 with 18F-Fluoride. Synlett 2015, 27, 25-28, 10.1055/s-0035-1560592.
- Hang Shi; Augustin Braun; Lu Wang; Steven H. Liang; Neil Vasdev; Tobias Ritter; Synthesis of 18F-Difluoromethylarenes from Aryl (Pseudo) Halides.. Angewandte Chemie International Edition 2016, 55, 10786-10790, 10.1002/anie.201604106.
- Gengyang Yuan; Feng Wang; Nickeisha A. Stephenson; Lu Wang; Benjamin H. Rotstein; Neil Vasdev; Pingping Tang; Steven H. Liang; Metal-free 18F-labeling of aryl-CF2H via nucleophilic radiofluorination and oxidative C-H activation.. Chemical Communications 2016, 53, 126-129, 10.1039/c6cc07913j.
- Jeroen B. I. Sap; Thomas Wilson; Choon Wee Kee; Natan Straathof; Christopher W. Am Ende; Paramita Mukherjee; Lei Zhang; Christophe Genicot; Véronique Gouverneur; Synthesis of 18F-difluoromethylarenes using aryl boronic acids, ethyl bromofluoroacetate and [18F]fluoride.. Chemical Science 2019, 10, 3237-3241, 10.1039/c8sc05096a.
- Laura Trump; Agostinho Lemos; Bénédicte Lallemand; Patrick Pasau; Joël Mercier; Christian Lemaire; André Luxen; Christophe Genicot; Late‐Stage 18 F‐Difluoromethyl Labeling of N‐Heteroaromatics with High Molar Activity for PET Imaging. Angewandte Chemie International Edition 2019, 58, 13149-13154, 10.1002/anie.201907488.
- Laura Trump; Agostinho Lemos; Jérôme Jacq; Patrick Pasau; Bénédicte Lallemand; Joël Mercier; Christophe Genicot; André Luxen; Christian Lemaire; Development of a General Automated Flow Photoredox 18F-Difluoromethylation of N-Heteroaromatics in an AllinOne Synthesizer. Organic Process Research & Development 2020, XXXX, XXX-XXX, 10.1021/acs.oprd.9b00442.
- Jian Rong; Ling Deng; Ping Tan; Chuanfa Ni; Yucheng Gu; Jinbo Hu; Radical Fluoroalkylation of Isocyanides with Fluorinated Sulfones by Visible-Light Photoredox Catalysis. Angewandte Chemie International Edition 2016, 55, 2743-2747, 10.1002/anie.201510533.
- Wei-Jun Fu; Xin Han; Mei Zhu; Chen Xu; Zhiqiang Wang; Baoming Ji; Xin-Qi Hao; Mao-Ping Song; Visible-light-mediated radical oxydifluoromethylation of olefinic amides for the synthesis of CF2H-containing heterocycles. Chemical Communications 2016, 52, 13413-13416, 10.1039/C6CC07771D.
- Guanglong Zou; Xuelin Wang; Visible-light induced di- and trifluoromethylation of N-benzamides with fluorinated sulfones for the synthesis of CF 2 H/CF 3 -containing isoquinolinediones. Organic & Biomolecular Chemistry 2017, 15, 8748-8754, 10.1039/C7OB02226C.
- Mei Zhu; Wei-Jun Fu; Zhiqiang Wang; Chen Xu; Baoming Ji; Visible-light-mediated direct difluoromethylation of alkynoates: synthesis of 3-difluoromethylated coumarins. Organic & Biomolecular Chemistry 2017, 15, 9057-9060, 10.1039/C7OB02366A.
- Mei Zhu; Weijun Fu; Wenbo Guo; Yunfei Tian; Zhiqiang Wang; Chen Xu; Baoming Ji; Visible-Light-Induced Radical Di- and Trifluoromethylation of β, γ-Unsaturated Oximes: Synthesis of Di- and Trifluoromethylated Isoxazolines. European Journal of Organic Chemistry 2019, 2019, 1614-1619, 10.1002/ejoc.201801790.
- Mei Zhu; Qingqing You; Rongxia Li; Synthesis of CF2H-containing oxindoles via photoredox-catalyzed radical difluoromethylation and cyclization of N-arylacrylamides. Journal of Fluorine Chemistry 2019, 228, 109391, 10.1016/j.jfluchem.2019.109391.
- Huan Sun; Yue Jiang; Ying-Sha Yang; Yun-Yun Li; Lin Li; Wen-Xuan Wang; Tao Feng; Zheng-Hui Li; Ji-Kai Liu; Synthesis of difluoromethylated 2-oxindoles and quinoline-2,4-diones via visible light-induced tandem radical cyclization of N-arylacrylamides. Organic & Biomolecular Chemistry 2019, 17, 6629-6638, 10.1039/c9ob01213c.
- Agostinho Luís Pereira Lemos; Laura Trump; Bénédicte Lallemand; Patrick Pasau; Joël Mercier; Christian Lemaire; Jean-Christophe M. Monbaliu; Christophe Genicot; André Luxen; Radical C–H 18F-Difluoromethylation of Heteroarenes with [18F]Difluoromethyl Heteroaryl-Sulfones by Visible Light Photoredox Catalysis. Catalysts 2020, 10, 275, 10.3390/catal10030275.


