 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wieslawa Lesniak | + 3778 word(s) | 3778 | 2021-01-08 07:27:02 | | | |
| 2 | Dean Liu | -1184 word(s) | 2594 | 2021-01-18 05:09:02 | | |
Video Upload Options
Epidermal differentiation relies on a highly coordinated program of gene expression. Epigenetic mechanisms, which commonly include DNA methylation, covalent histone modifications, and microRNA (miRNA) activity, modulate various stages of gene expression by altering chromatin accessibility and mRNA stability. Their involvement in epidermal differentiation is a matter of
intensive studies.
1. Introduction
The epidermis constitutes the surface of the skin and is built of specialized epithelial cells called keratinocytes that are organized in several layers: Basal, spinous, granular, and the outermost—corneal layer[1]. Keratinocytes originate from two pools of quiescent epidermal stem cells (qESC), one residing in the basal layer of the interfollicular epidermis (IFE) and another in a so-called “bulge” situated in the hair follicle below the opening of the sebaceous gland; the bulge stem cells generate keratinocytes of the hair follicle (HF) lineage. Quiescent epidermal stem cells give rise to transient amplifying (TA) cells, which, in turn, generate “mature” keratinocytes that populate the basal epidermal layer or the outer root sheath of the hair follicle[2]. After an initial round of cell divisions, keratinocytes in the basal layer are pushed upward by newly generated cells, lose contact with the basal membrane, cease to proliferate and enter the differentiation phase. During differentiation, keratinocytes execute a highly ordered and time-synchronized synthesis of specific lipids and proteins necessary to build the cornified envelope, that is, a specialized sub-membranous structure, which provides a tight and resilient barrier against environmental hazards. This process is coupled with changes in keratinocyte morphology, i.e., the cells flatten, enucleate, and, when they reach the uppermost corneal layer, successively desquamate[1]. Thus, within about the 30 days necessary to complete this cycle in humans, keratinocytes pass through quiescence, transient proliferation stage, differentiation phase, and, finally, cell death (Figure 1A). Epidermal growth and differentiation entail a well-coordinated expression of multiple genes, which is orchestrated by various mechanisms of transcriptional and post-transcriptional control. This strict control is often challenged by external stressors (e.g., UV light, wounding), modulated by physiological factors (e.g., nutrient supply, aging), or dysregulated in skin diseases.
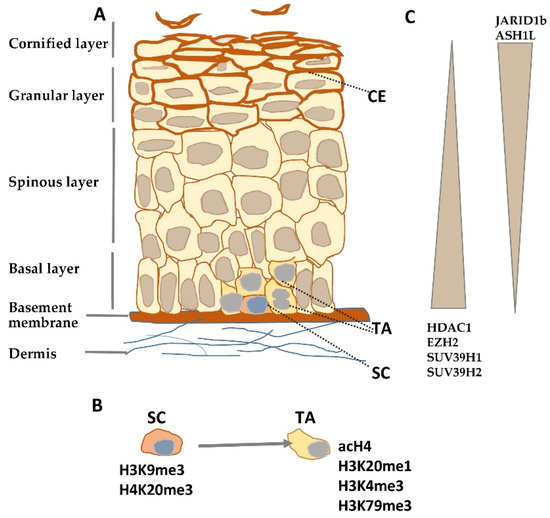
Figure 1. Histone modifying enzymes and histone modifications in epidermal differentiation. (A) Schematic representation of human epidermis. (B) Histone modifications typical for epidermal stem (SC) and transient amplifying (TA) cells. (C) Gradient of histone modifying enzyme expression in epidermis. CE—cornified envelope.
Genetic information, common to all individuals of a given species, is processed and modified by epigenetic mechanisms with the result that each individual cell or tissue acquires a different phenotype[3]. Epigenetic factors regulate gene expression at both the transcriptional and post-transcriptional levels and are especially important in developmental processes and cellular differentiation. DNA methylation/demethylation and histone modifications (e.g., methylation, acetylation, phosphorylation) alter chromatin structure and accessibility to the transcriptional machinery by facilitating or impeding the binding of chromatin-modifying complexes. On the other hand, microRNAs (miRNAs) induce cleavage of mRNAs or interfere with their translation. The extent of the contribution of epigenetic mechanisms to the control over epidermal differentiation is a subject of intensive studies. This review article offers an insight into how epigenetic factors contribute to and regulate keratinocyte differentiation.
2. Epigenetic Mechanisms Involved in Epidermal Differentiation
2.1. Histone Modifications
Histones are basic proteins that bind to DNA and organize the chromatin. Dimers of four core histones (H2A, H2B, H3, and H4) form an octamer, which, together with a 146 bp long fragment of DNA strand, constitutes the elementary chromatin unit, i.e., the nucleosome. In contrast, histone H1 binds to the internucleosomal (linker) DNA, stabilizing, and compacting the nucleosome structure[4]. The N-terminal histone tails, rich in basic amino acid residues, are subject to many covalent modifications, most of which (e.g., acetylation, methylation, phosphorylation, or ubiquitination) are reversible. These modifications, often referred to as a histone code, are catalyzed by specific groups of enzymes that include histone acetyltransferases (HATs), deacetylases (HDACs), methyltransferases (HMTs), and demethylases (HDMs). HATs and HDACs usually have broad specificity as to the histone type and amino acid position while HMTs and HDMs are more specific and usually methylate/demethylate a particular histone at a particular position and to a particular extent (e.g., mono-, di- or trimethylation of a lysine residue)[5][6]. Certain histone modifications, such as H3K4me3 H3K79me2/3, and H3K36me3 are found on promoters or gene bodies of actively transcribed genes and are often referred to as activating histone marks. Acetylation of lysine residues also promotes transcription since it neutralizes their positive charge and disrupts electrostatic interactions between histones and DNA, which results in less effective local DNA packaging[5]. On the other hand, H3K9me2/3, H3K27me2/3 and H4K20me3 and histone deacetylation are repressive histone modifications found on promoters of transcriptionally inactive genes. The activating or repressive effect on transcription of these or other histone modifications is exerted via a chain of protein interactions that ultimately engage ATP-dependent chromatin remodeling complexes. More specifically, modified histone residues serve as recognition sites for non-histone proteins endowed with specific modules e.g., so-called bromodomains, that can bind to various acetylated lysine residues, or chromodomains, that recognize particular methylated lysine residues with high specificity. These domains are present either in proteins (e.g., HATs) that are components of larger, chromatin-remodeling complexes containing, as a catalytic component, ATP-dependent RNA/DNA helicases, or in proteins, such as Brg1[7] or the SWI/SNF family remodelers, that are RNA/DNA helicases themselves[8]. There are 4 families of chromatin-remodeling complexes with slightly different functionalities; all of them rely on ATP-derived energy to slide or remove the nucleosome to expose cis-elements important for transcription and/or to modulate the arrangement and stability of nucleosomes towards either higher chromatin compaction or relaxation[8].
Histone modifications constitute a crucial factor in skin development and morphogenesis as it has been documented by studies on embryonic ablation of histone-modifying enzymes such as deacetylases[9] or methyltransferases[10]. Numerous studies certify that they also shape the architecture and functionality of the adult epidermis by controlling epidermal growth and differentiation. Quiescent stem cells residing in the interfollicular epidermis possess a characteristic pattern of histone modifications. Namely, they were found to be enriched in inhibitory histone marks, H3K9me3 and H4K20me3, and to have a low level of activating modifications, such as acetylated histone H4 (acH4) and histone H4 monomethylated on lysine 20 (H4K20me1) (Figure 1B)[11]. Upon overexpression of c-Myc, a transcription factor that, under physiological conditions, induces the transition from quiescent to TA cells[12], the level of H3K9me3 was diminished while that of acH4 was increased, and there was, albeit transient, an increase in the H4K20me1 level[11]. A rise in the level of activating histone modifications, H3K4me3 and H3K79me3, was also observed at this stage in the hair follicle stem cells[13].
Results of experiments performed on mice with conditional knockout/knockdown or overexpression of histone-modifying enzymes in basal, keratin-14 (K14) expressing keratinocytes, and those obtained using cultured keratinocytes with altered expression of these enzymes, largely substantiate the observed changes in histone modification pattern occurring at the early stage of epidermal growth. For example, the loss of the Setd8 methyltransferase, which monomethylates H3K20, resulted in a lower level of this activating histone mark and led to inhibition of progenitor cell proliferation in the basal layer, which, in consequence, impaired epidermal differentiation [10]. Setd8 deletion coincided with a diminished level of p63, a transcription factor that controls the balance between keratinocyte proliferation and differentiation by maintaining the proliferative potential of epidermal stem cells[14]. Knockout of Ezh2, encoding H3K27 methyltransferase, in the basal cells of neonatal mouse epidermis resulted in a lower proliferation rate and higher expression of epidermal differentiation markers, such as loricrin and filaggrin, synthesized by differentiating keratinocytes[15]. In this case, reduced proliferation of basal keratinocytes could be largely attributed to the de-repression of genes encoding cell cycle inhibitors, including p16INK4a. It was also found that promoters of many differentiation-specific genes bear the H3K27me3 inhibitory histone mark in undifferentiated keratinocytes and that the premature differentiation was caused by loss of this mark due to Ezh2 knockout[15]. Interestingly, the effect of Ezh2 knockout was very similar to that of Jmjd3, a H3K27 demethylase, overexpression, which provoked induction of genes encoding differentiation markers, such as keratin 1, keratin 10, S100A8, and involucrin, in primary human keratinocytes[16]. Conversely, siRNA-induced silencing of Jmjd3 expression blocked the induction of differentiation genes that were marked by the presence of H3K27me3 on their promoters[16]. Knockout of Suv39h1, a gene encoding histone methyltransferase that imposes another inhibitory histone mark, H3K9me3, in HaCaT cells, a stable keratinocyte-derived cell line, also resulted in induction of differentiation-associated genes encoding keratin 10, desmoglein 1, S100A8, and late cornified envelope (LCE1) proteins[17]. In addition, a naturally occurring mutation in Suv39h2 that results in enzyme inactivity provoked higher expression of genes encoding differentiation markers[18].
Depletion of histone deacetylases, Hdac1 and Hdac2, in the basal layer of mouse epidermis resulted in increased histone acetylation and enhanced keratinocyte proliferation leading to hyperplasia and thicker epidermis, although it also caused apoptosis and hair follicle dystrophy[19]. Silencing of Jarid1b, an enzyme, which demethylates H3 on lysine K4, reduced the level of the activating histone mark, H3K4me3, and led to diminished expression of differentiation markers in HaCaT cells, while overexpression enhanced differentiation and inhibited proliferation[20]. Studies on mice bearing a mutation that decreases transcription and expression of Ash1l, the gene that encodes an H3K36 methyltransferase, showed that adult animals had hyperproliferative epidermis with a more diffused localization of keratin 1 and keratin 14; the latter one, a marker of undifferentiated keratinocytes, being also present in the suprabasal layer[21]. The effect of knockout/down or overexpression of histone-modifying enzymes on epidermal growth/differentiation is summarized in Table S1, and changes in their expression during epidermal differentiation are illustrated in Figure 1C.
The above studies show that the control of epidermal differentiation by histone modifications is exerted over at least two important stages. Namely, histone modifications regulate the switch between epidermal stem cell quiescence and proliferation, as indicated by Sen and coworkers[16], but also the expression of epidermal differentiation genes. In the first case, the control is maintained over cell cycle inhibitor genes and/or transcription factors and pathways that supervise the transition between quiescence and proliferation. In the second case, inhibitory histone modifications are directly imposed on promoters of differentiation-induced genes in undifferentiated keratinocytes. Interestingly, as noted by several reports, the depletion of a particular histone-modifying enzyme did not cause a general induction or inhibition of differentiation-associated genes, which suggests that various histone modifications control the expression of individual genes or group of genes but not the whole differentiation program[15][16][17].
2.2. DNA Methylation
DNA methylation (addition of a methyl group to C-5 position of the cytosine ring in CpG dinucleotides) is another process essential for proper epidermal differentiation. DNA methyltransferase 1 (Dnmt1), is abundantly expressed in embryonic mouse epidermis[22]] but, after birth, Dnmt1-positive staining becomes limited to the basal epidermal layer containing proliferating keratinocytes, which parallels Dnmt1 localization observed in adult human and mouse epidermis[22][23]. Dnmt1 knockdown in primary human keratinocytes led to the loss of progenitor cells and premature differentiation in epidermal xenografts, which coincided with higher expression of Cdk inhibitors, e.g., p16INK4a[23]. These findings were largely reproduced in mice, in which epidermal deletion of Dnmt1 caused multiple alterations in hair growth associated mainly with reduced proliferation of TA cells[22]. The involvement of DNA methylation in this decisive step determining the fate of keratinocyte precursors is substantiated by results showing differences in DNA methylation, at single CpG resolution, between qHF-SC isolated in the telogen phase of hair growth, when stem cells are quiescent, and those isolated in anagen, when stem cells are activated to proliferate[24]. Furthermore, Dnmt1 knockdown in keratinocytes resulted in higher expression of differentiation genes[23]. These findings led to the conclusion that DNA methylation by DNMT1 sustains progenitor proliferation by blocking the expression of cell cycle inhibitors and represses keratinocyte differentiation[23].
Changes in DNA methylation occurring during keratinocyte differentiation have been evaluated by several studies. Analysis of DNA methylation in undifferentiated and differentiated primary human keratinocytes, performed using MeDIP microarrays encompassing promoter sequences of nearly 25,000 genes, showed significant demethylation in 232 differentiation-induced gene promoters[23]. This analysis, which pointed to a substantial influence of DNA methylation/demethylation on the process of epidermal differentiation, has been recently challenged by a study analyzing changes in methylation of 850,000 CpGs during differentiation of primary human keratinocytes[25]. Results of the latter analysis revealed a stable methylation landscape and a lack of correlation between the methylation status and gene expression level. This finding was in agreement with earlier studies focused on genes of the Epidermal Differentiation Complex (EDC), which maintained their methylation status throughout the differentiation of primary keratinocytes despite large changes in expression[26][27]. This may indicate that the impact of DNA methylation on epidermal differentiation is exerted at the early stage, probably deciding on the balance between proliferation/quiescence of progenitor cells, with a lesser effect on gene expression in keratinocytes already poised for differentiation.
2.3. MicroRNAs
MiRNA expression in the skin and differences in the level of particular miRNAs at different stages of epidermal biogenesis, starting from the embryonic stage, were described soon after recognition of the role of miRNAs in gene expression regulation in higher organisms[28]. The essential role of miRNAs in skin biogenesis was revealed by studies on the consequences of epidermal-specific deletion of genes encoding miRNA processing enzymes Dicer and/or Drosha[28][29]. The resulting inhibition of miRNA generation had a profound effect on mouse skin morphogenesis, especially affecting the development of hair follicles, due to reduced proliferation of hair follicle keratinocytes and loss of progenitors, while the interfollicular epidermis largely preserved its morphology and proliferation potential[28]. In line with this finding, other studies provided evidence of differential expression of miRNAs in epidermal and hair follicle lineages[30], which may be indicative of differences in the epigenetic control in these two populations of epidermal progenitor cells, at least when miRNAs are concerned. The pronounced effect of Dicer knockout on hair follicle development correlated with the reduction in Wnt signaling, which is essential for hair follicle formation through securing proper communication between the dermis and epidermis[31]. Furthermore, the pattern of miRNAs expression in hair follicles was shown to be different in telogen and anagen, pointing to the role of miRNAs in hair cycle control[32][33].
Regarding keratinocyte differentiation, several studies documented the pattern of changes in miRNA expression during the course of human keratinocyte differentiation in vitro and in vivo[34][35][36]. There is a number of reports documenting, through both knockout/knockdown and overexpression of individual miRNAs, their role in epidermal development and differentiation. For example, the miR-184 knockout mouse model displayed epidermal hyperplasia, combined with increased p63 expression, while overexpression of miR-184 induced hypoplasia and enhanced Notch signaling, without any obvious effect on the hair follicle[37]. As indicated earlier, p63 is a transcription factor that maintains the proliferative potential of epidermal stem cells, while Notch signaling promotes keratinocyte differentiation[38]. Although miR-184 was shown not to target p63 directly, there are other miRNAs that regulate p63 level, among them miR-203, whose expression is inversely correlated with that of p63 along the path of keratinocyte differentiation[36][39]. Another miRNA, miR-214, was shown to target β-catenin and cause lower activity of the Wnt pathway[32]. Overexpression of miR-214 in a mouse model caused a reduction in epidermal thickness and lower keratinocyte proliferation rate as well as hair follicle loss. These examples suggest that miRNAs are involved in the control of epidermal fate at the critical point between proliferation and differentiation. The role of numerous other miRNAs in skin morphogenesis and epidermal differentiation has been described in some recent review articles [36][40].
References
- Simpson, C.L.; Patel, D.M.; Green, K.J. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 565–580.
- Blanpain, C.; Fuchs, E. Epidermal stem cells of the skin. Annu. Rev. Cell. Dev. Biol. 2006, 22, 339–373.
- Biemont, C. From genotype to phenotype. What do epigenetics and epigenomics tell us? Heredity 2010, 105, 1–3.
- Wolffe, A.P. Transcriptional regulation in the context of chromatin structure. Essays Biochem. 2001, 37, 45–57.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521.
- Trotter, K.W.; Archer, T.K. The BRG1 transcriptional coregulatory. Nucl. Recept. Signal. 2008, 6, e004.
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304.
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818.
- Driskell, I.; Oda, H.; Blanco, S.; Nascimento, E.; Humphreys, P.; Frye, M. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J. 2012, 31, 616–629.
- Frye, M.; Fisher, A.G.; Watt, F.M. Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation. PLoS ONE 2007, 2, e763.
- Arnold, I.; Watt, F.M. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr. Biol. 2001, 11, 558–568.
- Lien, W.H.; Guo, X.; Polak, L.; Lawton, L.N.; Young, R.A.; Zheng, D.; Fuchs, E. Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 2011, 9, 219–232.
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 2017, 75, 1179–1190.
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135.
- Sen, G.I.; Weber, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 2008, 1865–1870.
- Sobiak, B.; Leśniak, W. Effect of SUV39H1 histone methyltransferase knockout on expression of differentiation-associated genes in HaCaT keratinocytes. Cells 2020, 9, 2628.
- Bannoehr, J.; Balmer, P.; Stoffel, M.H.; Jagannathan, V.; Gaschen, V.; Kuhni, K.; Sayar, B.; Drogemuller, M.; Howald, D.; Wieber, D.J.; et al. Abnormal keratinocyte differentiation in the nasal planum of labrador retrievers with hereditary nasal parakeratosis (HNPK). PLoS ONE 2020, 15, e0225901.
- Hughes, M.W.; Jiang, T.X.; Lin, S.J.; Leung, Y.; Kobielak, K.; Widelitz, R.B.; Chuong, C.M. Disrupted ectodermal organ morphogenesis in mice with a conditional histone deacetylase 1, 2 deletion in the epidermis. J. Investig. Dermatol. 2014, 134, 24–32.
- Sun, X.; Li, Z.; Niu, Y.; Zhao, L.; Huang, Y.; Zhang, S.; Chen, T.; Fu, T.; Yang, T.; An, X.; et al. Jarid1b promotes epidermal differentiation by mediating the repression of Ship1 and activation of the AKT/Ovol1 pathway. Cell Prolif. 2019, 52, e12638.
- Li, G.; Ye, Z.; Shi, C.; Sun, L.; Han, M.; Zhuang, Y.; Xu, T.; Zhao, S.; Wu, X. The histone methyltransferase Ash1l is required for epidermal homeostasis in mice. Sci. Rep. 2017, 7, 45401.
- Li, J.; Jiang, T.X.; Hughes, M.W.; Wu, P.; Yu, J.; Widelitz, R.B.; Fan, G.; Chuong, C.M. Progressive alopecia reveals decreasing stem cell activation probability during aging of mice with epidermal deletion of DNA methyltransferase 1. J. Investig. Dermatol. 2012, 132, 2681–2690.
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567.
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647.
- Smits, J.P.H.; Dirks, R.A.M.; Qu, J.; Oortveld, M.A.W.; Brinkman, A.B.; Zeeuwen, P.L.J.M.; Schalkwijk, J.; Zhou, H.; Marks, H.; van den Bogaard, E.H. Terminal keratinocyte differentiation in vitro is associated with a stable DNA methylome. Exp. Dermatol. 2020.
- Sobiak, B.; Graczyk-Jarzynka, A.; Leśniak, W. Comparison of DNA methylation and expression pattern of S100 and other epidermal differentiation complex (EDC) genes in differentiating keratinocytes. J. Cell. Biochem. 2016, 117, 1092–1098.
- Sobiak, B.; Leśniak, W. The effect of single CpG demethylation on the pattern of DNA-protein binding. Int. J. Mol. Sci. 2019, 20, 914.
- Andl, T.; Murchison, E.P.; Liu, F.; Zhang, Y.; Yunta-Gonzalez, M.; Tobias, J.W.; Andl, C.A.; Seykora, J.T.; Hannon, G.J.; Millar, S.E. The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr. Biol. 2006, 16, 1041–1049.
- Teta, M.; Choi, Y.S.; Okegbe, T.; Wong, G.; Tam, O.H.; Chong, M.M.; Seykora, J.T.; Nagy, A.; Littman, D.R.; Andl, T.; et al. Inducible deletion of epidermal Dicer and Drosha reveals multiple functions for miRNAs in postnatal skin. Development 2012, 139, 1405–1416.
- Yi, R.; O’Carroll, D.; Pasolli, H.A.; Zhang, Z.; Dietrich, F.S.; Tarakhovsky, A.; Fuchs, E. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat. Genet. 2006, 38, 356–362.
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008029.
- Ahmed, M.I.; Alam, M.; Emelianov, V.U.; Poterlowicz, K.; Patel, A.; Sharov, A.A.; Mardaryev, A.N.; Botchkareva, N.V. MicroRNA-214 controls skin and hair follicle development by modulating the activity of the Wnt pathway. J. Cell. Biol. 2014, 207, 549–567.
- Mardaryev, A.N.; Ahmed, M.I.; Vlahov, N.V.; Fessing, M.Y.; Gill, J.H.; Sharov, A.A.; Botchkareva, N.V. Micro-RNA-31 controls hair cycle-associated changes in gene expression programs of the skin and hair follicle. FASEB J. 2010, 24, 3869–3881.
- Hildebrand, J.; Rütze, M.; Walz, N.; Gallinat, S.; Wenck, H.; Deppert, W.; Grundhoff, A.; Knott, A. A comprehensive analysis of microRNA expression during human keratinocyte differentiation in vitro and in vivo. J. Investig. Dermatol. 2011, 131, 20–29.
- Song, Z.; Liu, D.; Peng, Y.; Li, J.; Zhang, Z.; Ning, P. Differential microRNA expression profile comparison between epidermal stem cells and differentiated keratinocytes. Mol. Med. Rep. 2015, 11, 2285–2291.
- Lee, A.Y. The role of microRNAs in epidermal barrier. Int. J. Mol. Sci. 2020, 21, 5781.
- Nagosa, S.; Leesch, F.; Putin, D.; Bhattacharya, S.; Altshuler, A.; Serror, L.; Amitai-Lange, A.; Nasser, W.; Aberdam, E.; Rouleau, M.; et al. MicroRNA-184 Induces a Commitment Switch to Epidermal Differentiation. Stem Cell Rep. 2017, 9, 1991–2004.
- Panelos, J.; Massi, D. Emerging role of Notch signaling in epidermal differentiation and skin cancer. Cancer Biol. Ther. 2009, 1986–1993.
- Lena, A.M.; Shalom-Feuerstein, R.; Rivetti di Val Cervo, P.; Aberdam, D.; Knight, R.A.; Melino, G.; Candi, E. miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ. 2008, 15, 1187–1195.
- Schneider, M.R. MicroRNAs as novel players in skin development, homeostasis and disease. Br. J. Dermatol. 2012, 166, 22–28.

