 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Panagiotis Paliogiannis | + 5826 word(s) | 5826 | 2021-01-14 04:51:46 | | | |
| 2 | Bruce Ren | -21 word(s) | 5805 | 2021-01-15 03:33:38 | | |
Video Upload Options
Lung cancer is the leading cause of death for malignancy worldwide. Its molecular profiling has enriched our understanding of cancer initiation and progression and has become fundamental to provide guidance on treatment with targeted therapies. Testing the presence of driver mutations in specific genes in lung tumors has thus radically changed the clinical management and outcomes of the disease. Numerous studies performed with traditional sequencing methods have investigated the occurrence of such mutations in lung cancer, and new insights regarding their frequency and clinical significance are continuously provided with the use of last generation sequencing technologies.
1. Introduction
Lung cancer (LC) is one of the most incident malignancies worldwide. According to with the Global Cancer Observatory (GCO), more than 2,200,000 new cases and approximately 1,800,000 deaths were estimated in the world in 2020 [1]. Higher age-adjusted incidence rates were estimated in North America and Europe (32.6 and 29 cases, respectively, per 100,000 inhabitants), while higher mortality rates were observed in Europe (22.6 per 100.000); mortality rates were similar in North America and China [1]. The closeness between incidence and mortality rates shows that LC remains highly lethal, despite recent improvements in the prevention, screening, diagnosis, and clinical management of the disease.
Cigarette smoking is considered to be the main determinant of the distribution of LC cases among genders and populations. In the past few decades, LC has been reported to be more common among males who were most commonly smokers; nevertheless, recent reports describe a decline in LC cases in men and a consistent emerging increase in women, especially in young ages [2]. A report from the United States reported higher LC incidence in young women than young men [3], and this trend does not seem to exclusively depend on the changing patterns in cigarette smoking [2]. Other known risk factors for lung cancer include exposure to secondhand smoke, mineral, metal dust, and radon, as well as asbestos, which is the main risk factor for the development of malignant mesothelioma [4]. With the introduction of screening guidelines and decrease in tobacco use in the United States, the mortality rate for lung cancer has recently decreased by 48% in males and 23% in females [5], while a significant increase in the LC five-year survival rates by an average of 1.3% a year has been observed in China over the past 12 years [6].
From a pathological perspective, LC has been classically divided into two major classes: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). The latter comprises the most common histological subtypes, such as adenocarcinoma, squamous cell carcinoma, and large cell carcinoma. Squamous cell carcinoma was the most frequent NSCLC histotype until the 1980s, when it was superseded by adenocarcinoma [7], probably due to changes in the characteristics of cigarettes (increased puff volume and nitrate levels), and the aforementioned incidence increase in women who tend to be mostly affected by adenocarcinomas. NSCLC accounts for approximately 85% of the LC cases currently observed worldwide [8].
Treatment strategies and prognoses of NSCLC largely depend on the stage of the disease at diagnosis. Surgery is highly curative in early stages but totally ineffective in patients with metastasis; unfortunately, the latter comprise the greatest part of the cases at the time of diagnosis. In this subset of patients, the advent of targeted agents represents the most important innovation over the last years. The discovery of activating mutations of the epidermal growth factor receptor (EGFR) in patients with lung adenocarcinoma led to the development of a new family of biological agents, called tyrosine kinase inhibitors (TKIs) [9]. These medications have revolutionized the clinical management of patients harboring EGFR mutations, whose survival nearly doubled compared to standard chemotherapy [8]. The increased frequency of resistance to TKIs redimensioned initial enthusiasm and led to the progressive discovery of novel agents targeting different molecular alterations and pathways. Currently, it is estimated that up to 69% of advanced NSCLC patients have druggable mutations in numerous genes, including EGFR, anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS1), Kirsten rat sarcoma virus (KRAS), V-raf murine sarcoma oncogene homolog B1 (BRAF), MET, human epidermal growth factor receptor (HER2), and other genes [10]. Nevertheless, to date, only drugs targeting EGFR and BRAF mutations, and ALK or ROS1 rearrangements have been approved for clinical use. In particular, the third generation anti-EGFR osimertinib is currently recommended for patients with EGFR-mutated lung adenocarcinoma, alectinib, or brigatinib for those harboring ALK rearrangements, and crizotinib for those with ROS1 rearrangement. In addition, patients with tumors harboring BRAF V600E mutations can be treated with a combination of dabrafenib (anti-BRAF) plus trametinib (anti-MEK). The list will be surely expanded in the near future because intensive research is ongoing and better-designed medications, like larotrectinib for NRTK-altered cases, selpercatinib for RET-mutated cases, and others in KRAS-mutated cases, showed encouraging results in recent clinical trials [11][12][13]. Recent reports evidenced that the epidemiological patterns of these and other targetable genetic alterations vary considerably throughout the world and among different geographical areas, populations, genders, and individuals with different characteristics or habits.
2. Molecular Epidemiology of the Main Druggable Genes in LC
2.1. EGFR
EGFR, also known as RBB, ERBB1, and HER1, is a gene located in the short arm of chromosome 7. It encodes a transmembrane protein with a large extracellular component (four domains and ~620 amino acids) that primarily serves as ligand-binding sites and that is anchored by a short helical transmembrane domain to the intracellular tyrosine kinase domain (TKD) [14].
This protein is a member of a family of four structurally related tyrosine kinase receptors (RTKs) that includes human epidermal growth factor receptor 2 (ERBBB2/HER2), ERBB3/HER3, and ERBB4/HER4. EGFR, upon activation by specific ligands, homo- or hetero-dimerizes at the cell membrane and triggers the receptor’s intrinsic tyrosine kinase activity with the autophosphorylation of the tyrosine residues in the C-terminal domain of the protein itself [9]. This results in the downstream activation of other signaling proteins that are associated with several transduction cascades. These cascades include the mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) pathway, the PI3K-AKT-mTOR pathway, the JNK pathway, and others implicated in cell migration, adhesion, and proliferation pathways (Figure 1) [15].
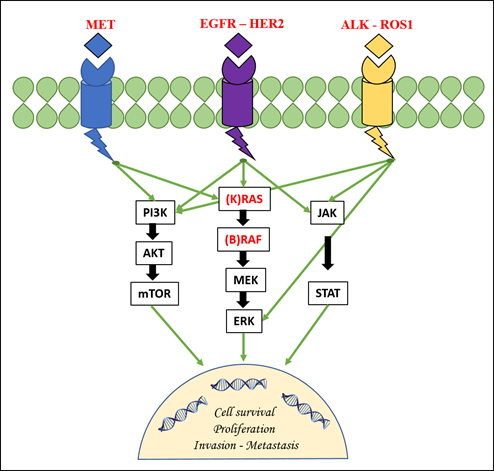
Figure 1. The figure depicts the main druggable genetic targets and their involvement in the main signaling pathways in non-small cell lung cancer. EGFR: epidermal growth factor receptor; HER2: human epidermal growth factor receptor 2; ERK: extracellular signal-related kinase.
Abnormalities within these pathways have been observed in NSCLC and are seen in tumors carrying mutations of the EGFR gene. In these neoplasms, EGFR plays a pivotal role in cellular proliferation, the inhibition of apoptosis, angiogenesis, and metastatic progression and chemoresistance. In NSCLC, EGFR mutations are limited to the first four exons (exons 18–21) of the tyrosine kinase domain, which encode the N-lobe and the 5′ portion of the C-lobe of EGFR; they consist of three different types of mutations (deletions, insertions, and missense point mutations), and they all cluster around the ATP binding pocket of the TKD of EGFR [16][17]. These mutations destabilize the autoinhibited conformation of the receptor, and the protein is then constitutionally activated to produce its pro-oncogenic effects [18]. On the other hand, these mutations are associated with hypersensitivity to TKIs, as we mentioned above, and are thus termed “sensitizing” EGFR mutations.
The prevalence of EGFR mutation varies with histotype, ethnicity, and other demographic or pathological factors. Several studies have shown that sensitizing EGFR mutations are almost exclusively observed in NSCLC patients with adenocarcinoma, rather than in those with other histologies; this observation is even more striking in East Asian populations, where EGFR mutations are present in up to 78% of adenocarcinomas, as opposed to only 10%–16% of adenocarcinomas in other ethnicities [18][19][20][21]. The reasons for such a discrepancy in mutational rates are unclear, with some studies suggesting alternative mechanisms that may be required for the development of lung cancer in Asians, who seem to have an inherent non-environmental susceptibility to the development of EGFR mutations [22][23]. Frequencies in Middle East and African populations are slightly higher than those observed in Western populations but still lower than in Asian populations. A slightly lower prevalence has been observed in the Oceanic ethnicities and other insular Mediterranean populations (12%) [24][25].
About 90% of NSCLC-specific sensitizing EGFR mutations are either in-frame microdeletions around the Leu–Arg–Glu–Ala (LREA) residues of exon 19 (known as Del19) or missense leucine-to-arginine substitution at codon 858 (L858R) in exon 21 [26][27]. Del19 alone, of which at least 30 variants have been identified, accounts for 45%–60% of all mutations, while L858R accounts for 35% to 45% of all mutations. Globally, these two types of mutations are termed “classic” or “common” EGFR mutations, and they are thought to produce similar configurational changes within the ATP binding cleft of the protein [16]. The remaining EGFR-mutant patients carry “uncommon” mutations. These comprise a heterogeneous group and have been found in 7%–23% of patients with EGFR mutant NSCLC [17], even though there are discrepancies across studies and their true individual frequency remains to be better determined. They can be isolated or occur together with an independent EGFR mutation (“complex” or “compound” mutations). The most frequently identified uncommon mutations are G719X, S768I, and L861Q in exons 18, 20, and 21, respectively. Larger studies performed with modern sequencing techniques have detected rarer mutations, such as A750P, T790M, L62R, S752F, Del18, and, more recently, even exon 18−25 kinase domain duplications and EGFR fusion events. Some studies have reported high frequencies (23%–43% of all uncommon mutations) of Ins20 mutations [17].
Regarding demographic factors, approximately all published studies have reported a higher frequency of EGFR mutations in women versus males, with figures of up to 69.7%. In other words, up to 42% of females versus only 14% of males with NSCLC are expected to harbor an EGFR TK domain mutation [21,28]. Regarding patient age at diagnosis, multiple targetable genomic alterations (including EGFR) have been found to be more prevalent in patients diagnosed at a younger age, although some controversial findings exist. Smoking status is another independent factor associated with the EGFR mutation incidence that is higher in never smokers (up to 66.6% of the cases) [28][29]. This was confirmed in a recent meta-analysis that included 167 epidemiologic studies with over 63,000 LC cases to calculate summary odds ratios for specific mutations (including EGFR) in never and ever smokers [30]. The meta-analysis also showed that as the smoking history increased, there were decreased odds for exhibiting the EGFR mutation, particularly for cases >30 pack-years. This is particularly true for Caucasian non-smokers, where the mutation prevalence seems to be relatively greater than in Asian non-smokers. Table 1 summarizes the main epidemiological data available regarding EGFR and the other driver genes discussed in the present review.
From a clinical point of view, tumor stage at diagnosis does not generally seem to correlate with the presence of EGFR mutations [31][32], but such mutations have been linked to the presence of multiple lung lesions and air bronchograms on imaging. Older data showed that ever-smokers with EGFR-mutated lung adenocarcinoma tend to be diagnosed with more advanced pathological stage disease.
Table 1. Main epidemiological features of molecular alterations in main candidate genes in non-small cell lung cancer.
|
Gene |
Most Common Variants |
Prevalence |
Age |
Gender |
Smoking |
Histology |
Prognostic Significance |
|
EGFR |
Mutations in exons 19 and 21 |
10%–16% in Western populations, 40%–50% in Asians. |
Younger patients |
Females |
Never smokers |
Adenocarcinoma |
Response to specific TKIs, T790M predictor of resistance |
|
ALK |
EML4-ALK variants |
1%–10% of NSCLC |
Younger patients |
Females? Not clear |
Never smokers |
Adenocarcinoma |
Aggressive tumors, response to specific TKIs |
|
ROS1 |
CD74-ROS1 variants |
0.9%–2.6% of NSCLC |
Younger patients |
Females |
Never smokers |
Adenocarcinoma |
Less aggressive tumors, response to specific TKIs |
|
KRAS |
Mutations in codons 12 and 13 |
30%–40% of NSCLC, more common in Caucasians |
Older ages |
Males? Not clear |
Smokers |
Adenocarcinoma |
Poor prognosis or response to TKIs? Not clear |
|
BRAF |
Mutations in exon 15 |
2%–4% of NSCLC |
No predilection |
V600E in females and others in males |
Smokers |
Adenocarcinoma |
Aggressive tumors and poor prognosis? Not clear. Response to BRAF inhibitors. |
|
MET |
Mutations in exon 14, amplification |
Mutations in 1%–10% of NSCLC, amplification in 5%–22% |
Older ages |
Not clear |
Smokers |
Adenocarcinoma and SCC |
Resistance to EGFR-TKIs. Response to MET inhibitors. |
|
HER2 |
Mutations in exons 18–21, amplification |
Mutations in 2%–3% and amplifications in 2%–5% of adenocarcinomas |
Not clear |
Mutations in females and amplifications in males |
Mutations in never smokers and amplifications in ex-smokers |
Adenocarcinoma Rare in SCC |
Resistance to EGFR-TKIs? Not clear |
NSCLC: non-small cell lung cancer; SCC: squamous cell carcinoma; TKIs: tyrosine kinase inhibitors; EML4: echinoderm microtubule-associated protein like 4;
As several randomized trials have extensively demonstrated, the presence of a classic sensitizing EGFR mutation is the most powerful predictive biomarker of response to monotherapy with first-generation TKIs and, thus, the most important prognostic factor in cases of advanced lung adenocarcinoma [33][34][35]. In this subset of cases, poorer outcomes have been associated with the male sex, the presence of the L858R mutation, and the diagnosis of the former bronchioalveolar adenocarcinoma. Exon 18 aberrations also appear to confer a poorer prognosis in relation to other EGFR mutations [36]. Despite the initial effectiveness of TKIs, all EGFR-mutated tumors eventually progress due to the occurrence of secondary mutations in the EGFR TKD or in genes involved in alternative molecular pathways. In about 50% of cases, it is the so-called gatekeeper T790M, also known as Thr790Met, that causes drug resistance by increasing the affinity of the domain for ATP [37][38]. T790M can also be present de novo, as a co-occurring mutation, in a small proportion of EGFR-positive patients, explaining primary resistance to first-generation TKIs. Second-generation irreversible TKIs target the acquired T790M mutation. A few other unclassical EGFR mutations are predictive of resistance to treatment with TKIs, especially exon 20 insertions [39][40][41].
2.2. ALK
The ALK gene consists of 30 exons mapping to the long arm of chromosome 2 (2p23.2–p23.1). It encodes a transmembrane protein of 1620 amino acids that belongs to the insulin receptor superfamily of RTKs, and it contains an extracellular domain, a transmembrane domain, and an intracellular domain [42]. ALK plays essential roles in the development of the brain, but it is also expressed in scattered adult cells such as neurons, glial cells, the testes, the pituitary gland, the hypothalamus, and endothelial cells [43]. Even though ALK is considered an orphan receptor, pleiotrophin and midkine are known ligands. Unlike for other receptors, the mechanisms for the activation of ALK are not fully understood. Triggering by the binding of the ligands to the extracellular domain seems the most likely, as well is the subsequent stimulation of classical signaling cascades such as MAPK, PI3K/mTOR, JAK-STAT, and SHH. In pathologic conditions, the downstream activity of these pathways leads to increased cell proliferation and metabolism, migration, survival, and apoptosis avoidance [43].
ALK aberrations in NSCLC were first identified in 2007. Most alterations are chromosomal rearrangements that result in the formation of fusion genes through small inversions within chromosome 2 [42]. The breakpoints for the translocations of ALK genes are typically located at exons 19–20 or exons 20–21. Fusion genes comprising echinoderm microtubule-associated protein-like 4 (EML4) and the ALK genes were the first identified, as well as the most frequent in NSCLC, accounting for about 80% of all fusion events [44]. Generally, the resulting chimera proteins contain the complete kinase domain and have a potent oncogenic effect due to the accelerated tyrosine kinase activity mediated by the fusion partner [45]. Several EML4-ALK fusion variants have been identified in NSCLC, differing in the breaking point between the two genes [46]. Variants 1, 2, and 3a/3b are the most common, accounting for more than 80% of all EML4-ALK variants, regardless of clinical characteristics such as gender, smoking status, histology, and stage [46][47]. Variant 1 alone accounts for about 60% of all EML4-ALK [47]. It consists of a simple fusion between exon 13 of EML4 and exon 20 of the ALK gene [48]. Rarer ALK fusion partners in NSCLC are TFG, KIF5B, KLC1, STRN, TPR, HIP1, GCC2, DCTN1, SQSTM1, LMO7, BIRC6, PHACTR1, and PTPN3 [44][45].
ALK aberrations act as oncogenic drivers in 1%–10% of NSCLC cases according to recent epidemiological studies [49], but the incidence rates of such aberrations are significantly variable among different populations. One study reported a higher prevalence of ALK-positive disease in Asian (22.0%), Pacific (10.8%), and Māori (6.9%) ethnicity patients in comparison with New Zealand Europeans (4.4%) [49]. By contrast, other studies have reported similar frequencies among different ethnicities [48][50]. A relatively high prevalence of ALK-positive cases (13%) was found in a selected population of Asian females with never/light smoking history and adenocarcinoma histology [51].
ALK translocations are more prevalent among individuals in their fourth or fifth decade of life, considerably younger than both patients with NSCLC (median age of 70 years) and patients with tumors harboring EGFR mutations (60–65 years) [49][52]. Colombino et al. found a significantly higher incidence of ALK rearrangements in patients with lung adenocarcinoma at less than 50 years of age in comparison to older individuals. This is in accordance with epidemiological data regarding other cancers known to harbor high rates of ALK rearrangements, such as anaplastic large cell lymphomas and neuroblastomas, and involve commonly children and young adults [51]. Nevertheless, ALK translocations have been found in both patients aged younger than 40 and older than 70 in approximately all the studies performed. Never or light smokers are also more likely to carry an ALK rearrangement; in published studies, up to 70%–80% of patients are never smokers, while no clear gender predilection has emerged to date [49][50][51].
Regarding histology, ALK rearrangements are typically detected in lung adenocarcinomas with solid growth predominance. In particular, data from cytomorphologic analysis positively associate extracellular mucin, cribriform structure, signet ring cells, and hepatoid cytology with the presence of ALK rearrangements [49]. Acinar growth and bronchioalveolar patterns are rarely documented in ALK-rearranged tumors. Despite their large predominance in adenocarcinomas, ALK rearrangements are also sporadically reported in lung squamous cell carcinoma (SCC). In addition, considering the difficulties in establishing the exact histotype in small biopsies, it is currently recommended to perform ALK testing in tumors of a potentially mixed (adenosquamous) histology, except in those with adenocarcinoma.
It has been evidenced that ALK-positive NSCLC has a highly aggressive clinical behavior and the tendency to be found at a higher clinical stage at diagnosis in comparison with wild-type patients. Numerous trials have showed that the presence of ALK rearrangements is a dominant predictive factor of response to treatments with TKIs. However, heterogeneous responses have been reported, and almost all ALK-rearranged tumors eventually experience resistance to crizotinib. The sequencing of resistant tumor DNA allowed for the identification of resistant ALK point mutations in 20%–40% of the cases[53]. The L1196M gatekeeper mutation (analogous with the EGFR T790M mutation) and the G1269A mutation are the most frequent in patients resistant to crizotinib, while the highly resistant G1202R mutation accounts for less than 10% of cases. Other concurrent somatic mutations have recently been identified (T1151R, R1192P, A1280V, and L1535Q) to confer strong resistance to ALK-TKIs [44]. In addition, EML4-ALK patients show lower response rates to platinum-based chemotherapy when compared to those with EGFR mutations [54]. The short translocation forms (variant 3 and others) seem to be characterized by a more aggressive oncological behavior and lower sensitivity to ALK-TKIs than cases with long forms (variant 1 and others). Second-generation ALK inhibitors that may overcome some of these resistances have been recently introduced in clinical practice.
2.3. ROS1
ROS1 is a gene located in chromosome 6q22 [55]. It encodes a sizable 2347-amino-acid-long transmembrane receptor with an extracellular N-terminal domain, a hydrophobic transmembrane region, an intracellular C-terminal tyrosine kinase domain, and a carboxy-terminal tail [56]. ROS1 shares many structural and functional similarities with the ALK protein, but it has a unique and large extracellular domain containing six repeat motifs with high homology to the extracellular matrix, as well as fibronectin type III-like sequence repeats, that resembles a cell adhesion molecule [57]. ROS1 functions as an orphan RTK receptor, with an unknown ligand. As a consequence, little is known about its function; it seems that it is able to directly couple extracellular adhesion-mediated events to intracellular signaling mediated by tyrosine phosphorylation [58]. Wild-type ROS1 activation has been implicated in the mesenchymal-to-epithelial transition and in cellular differentiation during the development of kidney, lung, small intestine, heart, and lung cancer. The dysregulation of the ROS1 kinase activity via the acquisition of certain gene fusions leads to the activation of the downstream cascades of several oncogenic pathways such as PI3K-Akt, mTOR, RAS-MAPK/ERK, VAV3, SHP-1 and SHP-2, which variably impact cell differentiation, proliferation, growth, and survival [59[60].
ROS1 gene fusions were first identified in the human glioblastoma cell line U-118 MGROS1 and, subsequently, in lung cancer in 2007 [61]. Their overall frequency in NSCLC ranges from 0.9% to 2.6%, with similar rates across Asia, Europe, and North America and a worldwide prevalence estimated as 1.9%. Thus far, several fusion patterns have been described. CD74-ROS1 was the first ROS1 rearrangement identified in NSCLC and is by far the most frequently reported, consisting of 32% of all ROS1 fusions [61]. Other common translocations include SLC34A2-ROS1 (~17%), tropomyosin 3 (TPM3-ROS1; ~15%), syndecan 4 (SDC4-ROS1; ~11%), ezrin (EZR-ROS1; ~6%), and FIG-ROS1 (~3%). Further rearrangements such as leucine-rich repeats and immunoglobulin-like domains 3 (LRIG3-ROS1), endoplasmic reticulum protein retention receptor 2 (KDELR2-ROS1), coiled-coil domain containing 6 (CCDC6-ROS1), Golgi-associated PDZ and coiled-coil motif-containing gene (GOPC-ROS1), moesin gene (MSN-ROS1), and clathrin heavy chain gene (CLTC-ROS1) occur in less than 1% of the cases [62].
ROS1 epidemiological patterns are very similar to those of ALK translocations, as it is more common in young individuals and never smokers, as confirmed in a recent meta-analysis [63]. Nevertheless, ROS1 rearrangements seem to involve more commonly female patients, resembling, in this case, the EGFR mutations. In addition, ROS1 fusions are more prevalent in adenocarcinomas than in other NSCLC histologies [62][63][64]. Interestingly, the cytomorphologic features (extracellular mucin, cribriform structure, signet ring cells, and hepatoid cytology) of adenocarcinomas harboring ROS1 translocations are very similar to those described in ALK-rearranged cases, suggesting that fusion genes in lung adenocarcinomas have specific identifiable morphological features.
ROS1 rearrangements are significantly more frequent in advanced NSCLC clinical stages [63], but they were associated with lower rates of extrathoracic disease, including brain metastases, at initial metastatic diagnosis [65]. Patients with ROS1-rearranged disease are expected to respond to TKIs as much as ALK-rearranged tumors, and crizotinib was granted full approval in 2016 [66][67]. As for other TKIs, however, patients ultimately develop drug resistance. The discrepancy in sensitivity to ALK/ROS TKIs among different ROS1 fusion patterns has been described, but the underlying mechanisms remain to be elucidated [57].
2.4. KRAS
The KRAS gene is located on the short arm of chromosome 12 (12p12.1) and encodes a guanosine triphosphatase (GTPase) that links cell surface receptors to signaling pathways such as rapidly accelerated fibrosarcoma (RAF)-MEK-ERK, PI3K-AKT-mTOR, and RALGDS-RA [68]. KRAS is a proto-oncogene and acts as a molecular on/off switch regulator to cell proliferation, maturation, and differentiation. At the cell level, binding of an appropriate ligand (i.e., EGFR, ALK, or MET) with the KRAS protein leads to its dimerization, phosphorylation, and, thus, activation. RAS phosphorylation is determined by a balance of pro-phosphorylating guanine nucleotide exchange factors (GEFs) and negatively regulating GTPase activating proteins (GAPs). Activated RAS can activate downstream effectors belonging to the previously mentioned pathways. KRAS is the most frequent oncogene in cancer [68]. KRAS mutations have been found in numerous malignancies, including gastric, biliary, skin, and gynecological cancer, and they are the most common genetic alterations in pancreatic, colorectal, and lung cancer [69][70][71][72][73]. In NSCLC, KRAS mutations occur in about one third of the cases, thus representing the second most common genetic alteration after p53 mutations [74][75]. The vast majority of activating KRAS mutations in NSCLC are found in codons 12 and 13 (rarely in codon 61) and change the amino acid glycine in the KRAS protein [76]. They include G12C, G12V, and G12D. These mutations result in the loss of intrinsic GTPase activity, the persistence of the GTP-bound state, and, consequently, the deregulation of cell proliferation signals [77].
In NSCLC, as opposed to EGFR mutations, KRAS mutations are more common in Western than in Asian or Australian populations (23%–33% vs. 2%–15%, respectively, of cases) and in long-term tobacco smokers than in never-smokers (20%–44% vs. 6%–10%, respectively) [78][79][80][81]. KRAS transition mutations (G → A) are more common in patients with adenocarcinoma and no smoking history, whereas transversion mutations (G → T or G → C) are associated with tobacco exposure. This observation suggests that KRAS-mutated NSCLC in non-smokers may not be caused by second-hand tobacco exposure [82]. Unlike EGFR aberrations, KRAS mutations do not seem to have any gender or age predilection, even though young women have been reported to display a higher susceptibility to the transversion mutation G12C. This mutation is the single most frequent mutation among smokers, suggesting an increased susceptibility to tobacco carcinogenesis in women compared to men [83]. In contrast, Colombino et al. found a significantly higher incidence of KRAS mutations in males in a recent study performed in Italy.
The prognostic impact of KRAS mutations in NSCLC is not clear. Several individual studies and a metanalysis of 28 studies showed that tumors harboring somatic KRAS mutations have a worse prognosis and a reduced or absent response to EGFR TKIs [84][85][86][87]. Nevertheless, these results were not confirmed in other studies; for example, in the JBR.10 randomized controlled trial, KRAS mutations were not predictive of survival (HR for OS: 1.23, 95% CI: 0.76–1.97) [88]. In addition, KRAS has not been yet targeted directly in lung cancer and attempts to inactivate either upstream and downstream proteins involved in RAS signaling have been met with limited success to date.
2.5. BRAF
BRAF is a gene mapping on chromosome 7q34 encoding a protein that is a member of the RAF kinase family, which comprises three serine-threonine kinases: ARAF, BRAF, and CRAF. The BRAF protein contains 766 amino acids and is arranged into three highly conserved regions called CR1, CR2, and CR3. CR1 is located near the N-terminus and comprises both a cysteine-rich domain and a Ras-binding domain, while CR2 links the CR1 and CR3 domains and contains a binding site for the 14-3-3 inhibitory protein. Finally, CR3, which resides close to the C-terminus, contains a kinase domain that phosphorylates and activates downstream targets [89]. In addition, CR1 interacts with CR2 to disable the activation of the protein, which requires the binding of Ras-GTPases at the cellular membrane. Once activated, the BRAF protein forms homodimers with itself and heterodimers with other RAF kinases, and it plays important roles in regulating the MAPK/ERK signaling pathway, ultimately inducing cell growth, mobility, and survival [90].
The oncogenic potential of BRAF mutations was first described in 2002 by Davies et al. in melanoma and NSCLC cell lines [91]. Subsequent studies showed that mutations in BRAF result in the constitutive activation of the MAPK/ERK signaling pathway, leading to uncontrolled cellular proliferation and cell survival. They have been detected in various tumors, including NSCLC. BRAF mutations are involved in approximately half the cases of malignant cutaneous melanomas, as well as in sinonasal and other melanomas to a lesser extent, and represent a paradigm of successful molecular targeting therapies [92][93]. Other cancers in which BRAF is recurrently mutated include papillary thyroid carcinoma, hairy cell leukemia, and colorectal cancer [94][95][96]. In NSCLC, BRAF mutations occur in 2%–4% of cases, with similar figures in Asian and Caucasian populations [96]. BRAF mutations are typically distributed throughout exons 11 and 15, with most of them occurring at exon 15, which encodes the catalytic domain of the kinase. They have been classified into three functional classes based on signaling mechanism, kinase activity, and sensitivity to inhibitors: RAS-independent kinase-activating V600 monomers (class I), RAS-independent kinase-activating dimers (class II), and RAS-dependent kinase-inactivating heterodimers (class III) [97][98]. Class I V600E mutations are the most predominant, as they account for 20%–30% of all BRAF mutations. They result in strong activation of the MAPK/ERK pathway, regardless of RAS, whose activation is suppressed through a negative feedback loop triggered by ERK activation, involvement. Class II (L597Q/R, G464V/A, G469A/V/R/S, K601E/N/T, E451Q, A712T, and fusions) mutations hold an intermediate kinase activity and RAS independence; they account for about 20% of all BRAF mutations. Class III (G469E, G466V/E/A, N581S/I, D594G/N, and G596R) mutations rely on RAS activation to overcome negative feedback from ERK [97][98].
Globally, BRAF mutations have a strong male (61%) and ever-smoker predominance (81%), with variable differences among different mutational classes [99]. Class I mutations, for instance, are more likely to occur in females, whereas males are more likely to harbor non-V600 mutations [97][98][100]. In addition, several studies have shown that class I mutations are more frequent among never-smokers in comparison with class II or III [98][101]. No correlation with age at diagnosis has been reported so far [100]. BRAF mutations are almost exclusively detected in adenocarcinomas, with only anecdotal reports in SCC [98][100][101].
Regarding prognosis, class I mutations appear to have a slightly better prognosis than their class II and III counterparts, which have been associated with a more aggressive behavior and less favorable clinical course, as well as earlier disease progression after first-line chemotherapy [101]. However, these findings were not confirmed in all studies, and whether BRAF mutational status actually affects oncological outcomes remains to be clarified. What is certain is that the presence of the BRAF V600 mutation in NSCLC is a predictive marker of response to specific RAF inhibitors, such as vemurafenib or dabrafenib, in monotherapy or in combination with a MEK inhibitor. In addition, it has been observed that MAPK alterations, including BRAF and SHP2 mutations, are more frequent in patients who respond to PD-1 checkpoint inhibitors like nivolumab and pembrolizumab [102]. This may depend on the fact that these mutations are more frequently observed in smokers with a higher mutational load and thus a greater benefit with immunotherapy. Non-V600 mutations are currently not eligible for targeted therapies; however, interdependence from RAS suggests that class III BRAF mutant tumors could be sensitive to RTK inhibitors.
2.6. MET
The MET gene, also known as hepatocyte growth factor receptor (HGFR) resides in the long arm of the chromosome 7 (7q21–q31) and encodes an RTK for the hepatocyte growth factor (HGF). After posttranslational cleavage and glycosylation, a 50-kDa alpha chain and a 140-kDa beta chain are produced. The alpha chain is linked to the extracellular portion of the beta chain, which contains the semaphoring domain, a plexin-semaphorin-integrin (PSI) domain, four immunoglobulin-plexin-transcription (IPT) repeats, a transmembrane domain, a juxtamembrane domain, a tyrosine kinase domain, and the C-terminus. Similarly to other previously described proteins, MET also dimerizes upon binding to HGF, resulting in the phosphorylation of the docking sites for adaptor proteins that activate the downstream signaling to pathways such as MAPK/ERK and PI3K/AKT [103]. The intracellular juxtamembrane domain of the protein is encoded by exon 14 and contains critical regulatory elements, including the direct binding site for the ubiquitin ligase that promotes MET protein degradation [104]. Mutations in the MET gene can cause exon 14 skipping, and the resulting mutant receptor demonstrates enriched signaling and oncogenic potential, due to loss of a portion of its juxta-membrane domain.
MET aberrations have been well-documented in multiple oncogenic processes. In NSCLC, the three main mechanisms of MET dysregulation are protein overexpression and gene amplification or mutation. Gene rearrangements have been reported anecdotally [103]. The reported prevalence of driver mutations in the splice site of MET that result in exon 14 skipping in NSCLC ranges from 1% to 10%, with lower figures among Asian compared to Western populations [105][106]. A consistently greater incidence of MET amplifications has been extensively documented as a mechanism of acquired resistance in 5%–22% EGFR-mutated NSCLC upon therapeutic pressure with EGFR-TKIs [105]. Nevertheless, the incidence of MET amplification is significantly lower, ranging from 2% to 5% in naïve NSCLC patients, as reported by Colombino et al. in a study performed on 1440 patients with adenocarcinoma [107][108]. This confirms that pharmacologic pressure acts as a triggering mechanism of MET amplification. MET amplification stimulates in vitro cell proliferation and migration, promoting the progression of the disease and metastases formation in EGFR-mutated NCSLC tumors [109]. In addition, MET gene amplification causes first-generation EGFR-TKI resistance by activating the EGFR-independent phosphorylation of ERBB3 and the downstream activation of the PI3K/AKT pathway, thus providing a bypass mechanism.
MET-mutated NSCLC has been correlated with older age at diagnosis compared with EGFR, KRAS, and BRAF-mutant lung cancers, with a median age of 72.5 years; furthermore, two-thirds of patients harboring MET exon 14 mutations have been reported to be current or former smokers, but in a recent Italian study, no differences in MET amplification in relation with the smoking status were found in naïve patients with lung adenocarcinoma. MET amplification predominately occurs in adenocarcinomas and also seems to have correlated with an earlier stage of the disease in some studies, but several authors have reported conflicting results [110]. In the study of Go et al. MET amplification occurred in 3.9% of the 180 enrolled NSCLC patients and were more common in SCC than in those with adenocarcinoma [111]. NSCLC-harboring MET alterations are sensitive to treatment with inhibitors such as crizotinib and cabozantinib, and the therapeutic effect of these drugs is proportionally correlated with the levels of amplification; indeed, patients with higher levels of MET amplification are also expected to have longer progression-free survival (PFS) [110]. Nevertheless, in naïve patients, a higher MET gene copy number has been associated with worst outcomes.
2.7. HER2
HER2 is a gene located at the long arm of chromosome 17. It encodes an RTK protein of the ERBB family. HER2 has the highest activity among the receptors of this family, has no known ligands but can directly modulate EGFR signaling by combining with one of all the other members of the ERBB family to activate downstream MAPK, P3K/Akt, and JAK-STAT signaling pathways [111]. HER2 alterations in NSCLC include gene mutations and amplifications. HER2 mutations and HER2 amplifications have been reported in 2%–3% and 2%–5% of lung adenocarcinomas, respectively [112]. Nevertheless, HER2 was found to be amplified in 12%–13% of NSCLC cases that have acquired resistance to EGFR-TKIs [113]. HER2 driver mutations are invariably restricted to the first four exons (exons 18–21) of the tyrosine kinase domain of the gene; they have striking similarities to EGFR alterations concerning mutation type and specific clinicopathological features [114][115]. The most commonly observed HER2 mutation is an in-frame 12-bp insertion in exon 20 at the identical corresponding region of EGFR. It induces cell proliferation, motility, and survival processes [116].
HER2 mutations are more prevalent in patients of Asian ethnicity than in other populations. A significant association between never-smoker status and female sex has been reported [117], while HER2 amplifications seem to be more common in males and former-smokers [118]. In addition, HER2 driver mutations and amplifications do not commonly overlap in the same patient, as no HER2-mutant tumors have been found to be amplified, which is a finding consistent with The Cancer Genome Atlas (TCGA) data. This evidence suggests that HER2 driver mutations actually represent distinct pathogenic entities and different molecular targets, and they should not be both referred to as “HER2-positive lung cancer. However, series of cases harboring both HER2 amplification and mutation have also been sporadically reported. The presence of a HER2 mutation correlates with adenocarcinoma subtype, and the mutation rate seems to differ among lung adenocarcinoma subtypes, being mostly observed in acinar predominant adenocarcinoma (APA), papillary predominant adenocarcinoma (PPA), minimally invasive adenocarcinoma (MIA), and invasive mucinous adenocarcinoma (IMA). There are also some sporadic reports of HER2 mutations in SCCs.
The impact of HER2 dysregulation in the prognosis of NSCLC remains controversial, with inconsistent data from small-sized studies. It has been suggested that the HER2 gene copy number may negatively affect sensitivity to anti-EGFR agents, being responsible for acquired resistance to EGFR-TKIs. Further studies are necessary to better elucidate the prognostic roles of different genetic alterations of HER2 in different subsets of naive and treated patients.
References
- Global Cancer Observatory. Available online: https://gco.iarc.fr (accessed on 30 December 2020).
- Fidler-Benaoudia, M.M.; Torre, L.A.; Bray, F.; Ferlay, J.; Jemal, A. Lung cancer incidence in young women vs. young men: A systematic analysis in 40 countries. Int. J. Cancer 2020, 147, 811–819.
- Jemal, A.; Miller, K.D.; Ma, J.; Siegel, R.L.; Fedewa, S.A.; Islami, F.; Devesa, S.S.; Thun, M.J. Higher lung cancer incidence in young women than young men in the United States. N. Engl. J. Med. 2018, 378, 1999–2009.
- Budroni, M.; Cossu, A.; Paliogiannis, P.; Palmieri, G.; Attene, F.; Cesaraccio, R.; Tanda, F. Epidemiology of malignant pleural mesothelioma in the province of Sassari (Sardinia, Italy). A population-based report. Multidiscip. Respir. Med. 2013, 8, 45.
- Siegel, R.L.; Miller, K.D.; Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Cheng, B.; Xiong, S.; Li, C.; Liang, H.; Zhao, Y.; Li, J.; Shi, J.; Ou, L.; Chen, Z.; Liang, P.; Liang, W.; He, J. An annual review of the remarkable advances in lung cancer clinical research in 2019. J. Thorac. Dis. 2020, 12, 1056–1069.
- Devesa, S.S.; Bray, F.; Vizcaino, A.P.; Parkin, D.M. International lung cancer trends by histologic type: Male:female differ-ences diminishing and adenocarcinoma rates rising. Int. J. Cancer 2005, 117, 294–299.
- Rolfo, C.; Castiglia, M.; Perez, A.; Reclusa, P.; Pauwels, P.; Sober, L.; Passiglia, F.; Russo, A. Liquid biopsy in Non-Small Cell Lung Cancer (NSCLC). In Liquid Biopsy in Cancer Patients, 3rd ed.; Giordano, A., Russo, A., Rolfo, C., Eds.; Springer Interna-tional Publishing AG: Cham, Switzerland, 2017; pp. 103–115, ISBN 978-3-319-55661-1.
- Paliogiannis, P.; Attene, F.; Cossu, A.; Defraia, E.; Porcu, G.; Carta, A.; Sotgiu, M.I.; Pazzola, A.; Cordero, L.; Capelli, F.; et al. Impact of tissue type and content of neoplastic cells of samples on the quality of epidermal growth factor receptor mutation analysis among patients with lung adenocarcinoma. Mol. Med. Rep. 2015, 12, 187–191.
- Wheeler, D.A.; Wang, L. From human genome to cancer genome: The first decade. Genome Res. 2013, 23, 1054–1062.
- Arbour, K.C.; Riely, G.J. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: A Review. JAMA 2019, 322, 764–774.
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.
- Singh, H.; Kinarivala, N.; Sharma, S. Multi-targeting anticancer agents: Rational approaches, synthetic routes and structure activity relationship. Anticancer Agents Med. Chem. 2019, 19, 842–874.
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768.
- Roskoski, R. Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74.
- Bae, N.C.; Chae, M.H.; Lee, M.H.; Kim, K.M.; Lee, E.B.; Kim, C.H.; Park, T.I.; Han, S.B.; Jheon, S.; Jung, T.H.; et al. EGFR, ERBB2, and KRAS mutations in Korean non-small cell lung cancer patients. Cancer Genet. Cytogenet. 2007, 173, 107–113.
- Masood, A.; Kancha, R.K.; Subramanian, J. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer harboring uncommon EGFR mutations: Focus on afatinib. Semin. Oncol. 2019, 46, 271–283.
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007, 11, 217–227.
- Tfayli, A.H.; Fakhri, G.B.; Al Assaad, M.S. Prevalence of the epidermal growth factor receptor mutations in lung adenocar-cinoma patients from the Middle East region. Ann. Thorac. Med. 2019, 14, 173–178.
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262.
- Zhang, Y.L.; Yuan, J.Q.; Wang, K.F.; Fu, XH.; Han, X.R.; Threapleton, D.; Yang, Z.Y.; Mao, C.; Tang, J.L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993.
- Sekine, I.; Yamamoto, N.; Nishio, K.; Saijo, N. Emerging ethnic differences in lung cancer therapy. Br. J. Cancer 2008, 99, 1757–1762.
- Tsao, A.S.; Tang, X.M.; Sabloff, B.; Xiao, L.; Shigematsu, H.; Roth J. Clinicopathologic characteristics of the EGFR gene muta-tion in non-small cell lung cancer. J. Thorac. Oncol. 2006, 1, 231–239.
- Benbrahim, Z.; Antonia, T.; Mellas, N. EGFR mutation frequency in Middle East and African non-small cell lung cancer pa-tients: A systematic review and meta-analysis. BMC Cancer 2018, 18, 891.
- Colombino, M.; Paliogiannis, P.; Cossu, A.; Santeufemia, D.A.; Sardinian Lung Cancer (SLC) Study Group; Sini, M.C.; Casula, M.; Palomba, G.; Manca, A.; Pisano, M.; et al. EGFR, KRAS, BRAF, ALK, and cMET genetic alterations in 1440 Sar-dinian patients with lung adenocarcinoma. BMC Pulm. Med. 2019, 19, 209.
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTI-MAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–744.
- Benlloch, S.; Botero, M.L.; Beltran-Alamillo, J.; Mayo, C.; Gimenez-Capitán, A.; de Aguirre, I.; Queralt, C.; Ramirez, J.L.; Ra-món y Cajal, S.; Klughammer, B.; et al. Clinical validation of a PCR assay for the detection of EGFR mutations in non-small-cell lung cancer: Retrospective testing of specimens from the EURTAC trial. PLoS ONE 2014, 9, e89518.
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346.
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967.
- Chapman, A.M.; Sun, K.Y.; Ruestow, P.; Cowan, D.M.; Madl, A.K. Lung cancer mutation profile of EGFR, ALK, and KRAS: Meta-analysis and comparison of never and ever smokers. Lung Cancer 2016, 102, 122–134.
- Marchetti, A.; Martella, C.; Felicioni, L.; Barassi, F.; Salvatore, S.; Chella, A.; Camplese, P.P.; Iarussi, T.; Mucilli, F.; Mezzetti, A.; et al. EGFR mutations in non-small-cell lung cancer: Analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J. Clin. Oncol. 2005, 23, 857–865.
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. Mutations of the epidermal growth factor re-ceptor gene in lung cancer: Biological and clinical implications. Cancer Res. 2004, 64, 8919–8923.
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Ki-noshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246.
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128.
- Cheng, C.; Wang, R.; Li, Y.; Pan, Y.; Zhang, Y.; Li, H.; Zheng, D.; Zheng, S.; Shen, X.; Sun, Y.; et al. EGFR exon 18 mutations in East Asian patients with lung adenocarcinomas: A comprehensive investigation of prevalence, clinicopathologic charac-teristics and prognosis. Sci. Rep. 2015, 5, 13959.
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.E.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J. Thorac. Oncol. 2013, 8, 179–178.
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M muta-tion in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075.
- Chen, M.; Xu, Y.; Zhao, J.; Zhong, W.; Zhang, L.; Bi, Y.; Wang, M. Concurrent driver gene mutations as negative predictive factors in epidermal growth factor receptor-positive non-small cell lung cancer. EBioMedicine 2019, 42, 304–310.
- Watanabe, M.; Kawaguchi, T.; Isa, S.; Ando, M.; Tamiya, A.; Kubo, A.; Saka, H.; Takeo, S.; Adachi, H.; Tagawa, T.; et al. Ul-tra-sensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR. Clin. Cancer Res. 2015, 2, 3552–3560.
- Chen, L.Y.; Molina-Vila, M.A.; Ruan, S.Y.; Su, K.Y.; Liao, W.Y.; Yu, K.; Ho, C.C.; Shih, J.Y.; Yu, C.J.; Yang, J.C.; et al. Coex-istence of EGFR T790M mutation and common activating mutations in pretreatment non-small cell lung cancer: A systematic review and meta-analysis. Lung Cancer 2016, 94, 46–53.
- Inamura, K.; Takeuchi, K.; Togashi, Y.; Nomura, K.; Ninomiya, H.; Okui, M.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Soda, M.; et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol. 2008, 3, 13–17.
- Rosas, G.; Ruiz, R.; Araujo, J.M.; Pinto, J.A.; Mas, L. ALK rearrangements: Biology, detection and opportunities of therapy in non-small cell lung cancer. Crit. Rev. Oncol. Hematol. 2019, 136, 48–55.
- Noh, K.W.; Lee, M.S.; Lee, S.E.; Song, J.Y.; Shin, H.T.; Kim, Y.J.; Oh, D.Y.; Jung, K.; Sung, M.; Kim, M.; et al. Molecular breakdown: A comprehensive view of anaplastic lymphoma kinase (ALK)-rearranged non-small cell lung cancer. J. Pathol. 2017, 243, 307–319.
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hat-anaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566.
- He, Y.; Sun, L.Y.; Gong, R.; Liu, Q.; Long, Y.K.; Liu, F.; Wang, F. The prevalence of EML4-ALK variants in patients with non-small-cell lung cancer: A systematic review and meta-analysis. Biomark. Med. 2019, 13, 1035–1044.
- Pan, Y.; Zhang, Y.; Li, Y.; Hu, H.; Wang, L.; Li, H.; Wang, R.; Ye, T.; Luo, X.; Zhang, Y.; et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: A comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer 2014, 84, 121–126.
- Takahashi, T.; Sonobe, M.; Kobayashi, M.; Yoshizawa, A.; Menju, T.; Nakayama, E.; Mino, N.; Iwakiri, S.; Sato, K.; Miyaha-ra, R.; et al. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann. Surg. Oncol. 2010, 17, 889–897.
- McKeage, M.J.; Tin Tin, S.; Khwaounjoo, P.; Sheath, K.; Dixon-McIver, A.; Ng, D.; Sullivan, R.; Cameron, L.; Shepherd, P.; Laking, G.R.; et al. Screening for Anaplastic Lymphoma Kinase (ALK) gene rearrangements in non-small cell lung cancer (NSCLC) in New Zealand. Intern. Med. J. 2020, 50, 716–725.
- Martelli, M.P.; Sozzi, G.; Hernandez, L.; Pettirossi, V.; Navarro, A.; Conte, D.; Gasparini, P.; Perrone, F.; Modena, P.; Pastorino, U.; et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am. J. Pathol. 2009, 174, 661–670.
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253.
- Hou, H.; Zhang, C.; Qi, X.; Zhou, L.; Liu, D.; Lv, H.; Li, T.; Sun, D.; Zhang, X. Distinctive targetable genotypes of younger patients with lung adenocarcinoma: A cBioPortal for cancer genomics data base analysis. Cancer Biol. Ther. 2020, 21, 26–33.
- Lovly, C.M.; Pao, W. Escaping ALK inhibition: Mechanisms of and strategies to overcome resistance. Sci. Transl. Med. 2012, 4, 120ps2.
- Takeda, M.; Okamoto, I.; Sakai, K.; Kawakami, H.; Nishio, K.; Nakagawa, K. Clinical outcome for EML4-ALK-positive pa-tients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann. Oncol. 2012, 23, 2931–2936.
- Nagarajan, L.; Louie, E.; Tsujimoto, Y.; Balduzzi, P.C.; Huebner, K.; Croce, C.M. The human c-ros gene (ROS) is located at chromosome region 6q16----6q22. Proc. Natl. Acad. Sci. USA 1986, 83, 6568–6572.
- Acquaviva, J.; Wong, R.; Charest, A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim. Biophys. Acta 2009, 1795, 37–52.
- Cai, W.; Li, X.; Su, C.; Fan, L.; Zheng, L.; Fei, K.; Zhou, C.; Manegold, C.; Schmid-Bindert, G. ROS1 fusions in Chinese pa-tients with non-small-cell lung cancer. Ann. Oncol. 2013, 24, 1822–1827.
- Chin, L.P.; Soo, R.A.; Soong, R.; Ou, S.H. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: A promising thera-peutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1625–1630.
- Pal, P.; Khan, Z. ROS1. J. Clin. Pathol. 2017, 70, 1001–1009.
- Charest, A.; Wilker, E.W.; McLaughlin, M.E.; Lane, K.; Gowda, R.; Coven, S.; McMahon, K.; Kovach, S.; Feng, Y.; Yaffe, M.B.; et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006, 66, 7473–7481.
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203.
- Zhang, Q.; Wu, C.; Ding, W.; Zhang, Z.; Qiu, X.; Mu, D.; Zhang, H.; Xi, Y.; Zhou, J.; Ma, L.; et al. Prevalence of ROS1 fusion in Chinese patients with non-small cell lung cancer. Thorac. Cancer 2019, 10, 47–53.
- Zhu, Q.; Zhan, P.; Zhang, X.; Lv, T.; Song, Y. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2015, 4, 300–309.
- Song, Z.; Yu, X.; Zhang, Y. Clinicopathological characteristics and survival of ALK, ROS1 and RET rearrangements in non-adenocarcinoma non-small cell lung cancer patients. Cancer Biol. Ther. 2017, 18, 883–887.
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis. Oncol. 2017, 1, 1–13.
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971.
- Sehgal, K.; Patell, R.; Rangachari, D.; Costa, D.B. Targeting ROS1 rearrangements in non-small cell lung cancer with crizo-tinib and other kinase inhibitors. Transl. Cancer Res. 2018, 7, S779–S786.
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465.
- Paliogiannis, P.; Cossu, A.; Tanda, F.; Palmieri, G.; Palomba, G. KRAS mutational concordance between primary and meta-static colorectal adenocarcinoma. Oncol. Lett. 2014, 8, 1422–1426.
- Polom, K.; Das, K.; Marrelli, D.; Roviello, G.; Pascale, V.; Voglino, C.; Rho, H.; Tan, P.; Roviello, F. KRAS mutation in gastric cancer and prognostication associated with microsatellite instability status. Pathol. Oncol. Res. 2019, 25, 333–340.
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A; Altana, M.; et al. Targeted therapies in cholangiocarcinoma: Emerging evidence from clinical trials. Medicina (Kaunas) 2019, 55, 42.
- Sini, M.C.; Doneddu, V.; Paliogiannis, P.; Casula, M.; Colombino, M.; Botti, G; Ascierto, P.A; Lissia, A.; Cossu, A.; Palmieri, G. Genetic alterations in main candidate genes during melanoma progression. Oncotarget 2018, 9, 8531–8541.
- Rodriguez-Freixinos, V.; Lheureux, S.; Mandilaras, V.; Clarke, B.; Dhani, N.C.; Mackay, H.; Butler, M.O.; Wang, L.; Siu, L.L.; Kamel-Reid, S.; et al. Impact of somatic molecular profiling on clinical trial outcomes in rare epithelial gynecologic cancer pa-tients. Gynecol. Oncol. 2019, 153, 304–311.
- Martin, P.; Leighl, N.B.; Tsao, M.S.; Shepherd, F.A. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J. Thorac. Oncol. 2013, 8, 530–542.
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- Forbes, S.; Clements, J.; Dawson, E.; Bamford, S.; Webb, T.; Dogan, A.; Flanagan, A.; Teague, J.; Wooster, R.; Futreal, P.A.; et al. COSMIC 2005. Br. J. Cancer 2006, 94, 318–322.
- Mills, N.E.; Fishman, C.L.; Rom, W.N.; Dubin, N.; Jacobson, D.R. Increased prevalence of K-ras oncogene mutations in lung adenocarcinoma. Cancer Res. 1995, 55, 1444–1447.
- Jiang, A.G.; Lu, H.Y. k-RAS mutations in non-small cell lung cancer patients treated with TKIs among smokers and non-smokers: A meta-analysis. Contemp. Oncol. (Pozn.) 2016, 20, 124–129.
- Sun, Y.; Ren, Y.; Fang, Z.; Li, C.; Fang, R.; Gao, B.; Han, X.; Tian, W.; Pao, W.; Chen, H.; et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J. Clin. Oncol. 2010, 28, 4616–4620.
- Smits, A.J.; Kummer, J.A.; Hinrichs, J.W.; Herder, G.J.; Scheidel-Jacobse, K.C.; Jiwa, N.M.; Ruijter, T.E.; Nooijen, P.T.; Looi-jen-Salamon, M.G.; Ligtenberg, M.J.; et al. EGFR and KRAS mutations in lung carcinomas in the Dutch population: Increased EGFR mutation frequency in malignant pleural effusion of lung adenocarcinoma. Cell. Oncol. (Dordr.) 2012, 35, 189–196.
- Fong, K.M.; Zimmerman, P.V.; Smith, P.J. KRAS codon 12 mutations in Australian non-small cell lung cancer. Aust. N. Z. J. Med. 1998, 28, 184–189.
- Riely, G.J.; Marks, J.; Pao, W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205.
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177.
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. As-sessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic re-view and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008, 9, 962–972.
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic path-ways. J. Thorac. Oncol. 2019, 14, 606–616.
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308.
- Mascaux, C.; Iannino, N.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart M.; Lothaire, P.; Meert, AP.; Noel, S.; Lafitte, J.J.; et al. The role of RAS oncogene in survival of patients with lung cancer: A systematic review of the literature with me-ta-analysis. Br. J. Cancer 2005, 92, 131–139.
- Winton, T.; Livingston, R.; Johnson, D.; Rigas, J.; Johnston, M.; Butts, C.; Cormier, Y.; Cormier, Y.; Goss, G.; Inculet, R.; Val-lieres, E.; et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N. Engl. J. Med. 2005, 352, 2589–2597.
- Sithanandam, G.; Kolch, W.; Duh, F.M.; Rapp, U.R. Complete coding sequence of a human B-raf cDNA and detection of B-raf protein kinase with isozyme specific antibodies. Oncogene 1990, 5, 1775–1780.
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Manca, A.; Paliogiannis, P.; Colombino, M.; Casula, M.; Lissia, A.; Botti, G.; Caracò, C.; Ascierto, P.A.; Sini, M.C.; Palomba, G.; et al. Mutational concordance between primary and metastatic melanoma: A next-generation sequencing approach. J. Transl. Med. 2019, 17, 289.
- Colombino, M.; Lissia, A.; Franco, R.; Botti, G.; Ascierto, P.A.; Manca, A.; Sini, M.C.; Pisano, M.; Paliogiannis, P.; Tanda, F.; et al. Unexpected distribution of cKIT and BRAF mutations among southern Italian patients with sinonasal melanoma. Derma-tology 2013, 226, 279–284.
- Abdullah, M.I.; Junit, S.M.; Ng, K.L.; Jayapalan, J.J.; Karikalan, B.; Hashim, O.H. Papillary thyroid cancer: Genetic alterations and molecular biomarker investigations. Int. J. Med. Sci. 2019, 16, 450–460.
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A; Kern, W.; Martelli, MP.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315.
- Palomba, G.; Doneddu, V.; Cossu, A.; Paliogiannis, P.; Manca, A.; Casula, M.; Colombino, M.; Lanzillo, A.; Defraia, E.; Pazzola, A.; et al. Prognostic impact of KRAS, NRAS, BRAF, and PIK3CA mutations in primary colorectal carcinomas: A population-based study. J. Transl. Med. 2016, 14, 292.
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj; A.; Berenguer, J.; Fernan-dez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF mutations classes I, II, and III in NSCLC patients included in the SLLIP trial: The need for a new pre-clinical treatment rationale. Cancers (Basel) 2019, 11, E1381.
- Lin, Q.; Zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H.; et al. The association be-tween BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J. Transl. Med. 2019, 17, 298.
- Couraud, S.; Barlesi, F.; Fontaine-Deraluelle, C.; Debieuvre, D.; Merlio, J.P.; Moreau, L.; Beau-Faller, M.; Veillon, R.; Mosser, J.; Al Freijat, F.; et al. Clinical outcomes of non-small-cell lung cancer patients with BRAF mutations: Results from the French Cooperative Thoracic Intergroup biomarkers France study. Eur. J. Cancer 2019, 116, 86–97.
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D.; et al. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat. Rev. 2018, 66, 82–94.
- Dagogo-Jack, I.; Martinez, P.; Yeap, B.Y.; Ambrogio, C.; Ferris, L.A.; Lydon, C.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; John-son, B.E.; et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant lung cancer. Clin. Cancer Res. 2019, 25, 158–165.
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Author correction: Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 1022.
- Salgia, R. MET in lung cancer: Biomarker selection based on scientific rationale. Mol. Cancer Ther. 2017, 16, 555–565.
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell. 2001, 8, 995–1004.
- Song, Z.; Wang, H.; Yu, Z.; Lu, P.; Xu, C.; Chen, G.; et al. De novo MET amplification in Chinese patients with non-small-cell lung cancer and treatment efficacy with Crizotinib: A multicenter retrospective study. Clin. Lung Cancer 2019, 20, e171–e176.
- Drilon, A.; Cappuzzo, F.; Ou, S.I.; Camidge, D.R. Targeting MET in lung cancer: Will expectations finally be MET? J. Thorac. Oncol. 2017, 12, 15–26.
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313.
- Kawakami, H.; Okamoto, I.; Okamoto, W.; Tanizaki, J.; Nakagawa, K.; Nishio, K. Targeting MET amplification as a new on-cogenic driver. Cancers (Basel) 2014, 6, 1540–1552.
- Baldacci, S.; Kherrouche, Z.; Cockenpot, V.; Stoven, L.; Copin, M.C.; Werkmeister, E.; Marchand, N.; Kyheng, M.; Tulasne, D.; Cortot, A.B. MET amplification increases the metastatic spread of EGFR-mutated NSCLC. Lung Cancer 2018, 125, 57–67.
- Awad; M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J. Clin. Oncol. 2016, 34, 721–730.
- Landi, L.; Cappuzzo, F. HER2 and lung cancer. Expert. Rev. Anticancer Ther. 2013, 13, 1219–1228.
- Li, X.; Zhao, C.; Su, C.; Ren, S.; Chen, X.; Zhou, C. Epidemiological study of HER-2 mutations among EGFR wild-type lung adenocarcinoma patients in China. BMC Cancer 2016, 16, 828.
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247.
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; John-son, B.E.; et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005, 65, 1642–1646.
- Song, Z.; Yu, X.; Shi, Z.; Zhao, J.; Zhang, Y. HER2 mutations in Chinese patients with non-small cell lung cancer. Oncotarget 2016, 7, 78152–78158.
- Mazières, J.; Peters, S.; Lepage, B.; Cortot, A.B.; Barlesi, F.; Beau-Faller, M.; Besse; B.; Blons, H.; Mansuet-Lupo, A.; Urban, T.; et al. Lung cancer that harbors an HER2 mutation: Epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol. 2013, 31, 1997–2003.
- Bu, S.; Wang, R.; Pan, Y.; Yu, S.; Shen, X.; Li, Y.; Sun, Y.; Chen, H. Clinicopathologic characteristics of patients with HER2 insertions in non-small cell lung cancer. Ann. Surg. Oncol. 2017, 24, 291–297.
- Li, B,T.; Ross, D.S.; Aisner, D.L.; Chaft, J.E.; Hsu, M.; Kako, S.L.; Kris, M.G.; Varella-Garcia, M.; Arcila, M.E. HER2 Amplifi-cation and HER2 mutation are distinct molecular targets in lung cancers. J. Thorac. Oncol. 2016, 11, 414–419.


