 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rosy Ghanem | + 1897 word(s) | 1897 | 2021-01-07 03:30:54 | | | |
| 2 | Bruce Ren | -21 word(s) | 1876 | 2021-01-14 03:40:49 | | |
Video Upload Options
The respiratory epithelium can be affected by many diseases that could be treated using aerosol gene therapy. Among these, cystic fibrosis (CF) is a lethal inherited disease characterized by airways complications, which determine the life expectancy and the effectiveness of aerosolized treatments. Beside evaluations performed under in vivo settings, cell culture models mimicking in vivo pathophysiological conditions can provide complementary insights into the potential of gene transfer strategies. Such models must consider multiple parameters, following the rationale that proper gene transfer evaluations depend on whether they are performed under experimental conditions close to pathophysiological settings. In addition, the mucus layer, which covers the epithelial cells, constitutes a physical barrier for gene delivery, especially in diseases such as CF. Artificial mucus models featuring physical and biological properties similar to CF mucus allow determining the ability of gene transfer systems to effectively reach the underlying epithelium.
1. Cystic Fibrosis and Current Protein Treatment
Cystic fibrosis (CF) is one of the most common inherited genetic disorders, affecting over 70,000 people worldwide [1]. CF is induced by numerous mutations listed in a single gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) [2]. The CFTR protein is a channel that participates in the exit of chloride ions from the cytoplasm. This channel also plays a key role in the regulation of another channel called ENaC (epithelial sodium channel), involved in the reabsorption of sodium. Altogether, those channels control the correct hydration of mucus on airway epithelial cells. Alteration or dysfunction of the CFTR channel leads thus to abnormal ion transport and mucus dehydration (Figure 1A). Such dehydrated sticky mucus is responsible for the diminution of mucociliary clearance (MCC), leading to a mucosal accumulation. This mucus stagnation provides a propitious environment for bacterial colonization and infections, inducing related chronic inflammation. In the CF context, polynuclear neutrophils accumulation in the mucus is responsible for the production and the excretion of lysosomal enzymes which directly contribute to tissue remodeling [3][4]. The remodeling of secretory cells induces an overproduction of mucin, the main constituent of mucus, leading to severe consequences for the respiratory exchanges [5].
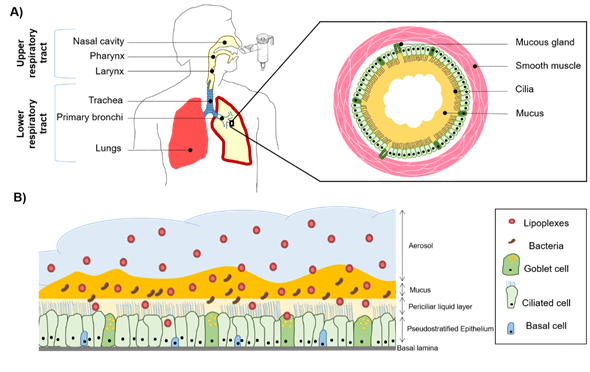
Figure 1. Schematic representation of the respiratory system and the use of an aerosol to target the pulmonary epithelium. (A) The upper and lower respiratory tract with an emphasis on a bronchial section constituted by mucous glands and smooth muscles. In the case of cystic fibrosis (CF), sticky mucus is overproduced and creates a bronchial plug. (B) Pseudostratified airway epithelium composed of goblet, ciliated, and basal cells. The mucosa is covered with airways surface liquid (ASL) which contains two layers, a periciliary liquid layer directly in contact with the cilia and mucus layer. CF mucus is colonized with opportunistic bacteria such as Staphylococcus aureus and Pseudomonas aeruginosa. These bacteria form a biofilm participating in the viscous and dense aspect of the CF mucus. When nanoparticles (NPs) such as lipoplexes are aerosolized, they have to cross the mucus barrier before reaching the epithelial cells to deliver therapeutic compounds such as acids nucleic construction.
Today, pulmonary complications are the primary cause of CF-related morbidity and mortality. Similar manifestations exist in the gastrointestinal and genital tracts but are, at present, dealt with by symptomatic treatments such as supplementary pancreatic enzymes [6]. During the last decade, new drugs have been developed regarding the CFTR mutations. Ivacaftor (Kalydeco®), a CFTR potentiator channel, is indicated for the G551D mutation. This potentiator can be associated with correctors such as lumacaftor (Orkambi®) or tezacaftor (Symdeko®) for the homozygote F508del mutation. More recently, a tritherapy, namely, elexacaftor-tezacaftor-ivacaftor (Trikafta®), showed an FEV1 (forced expiratory volume) higher than 10% benefit for patients with a single F508del allele [7]. Unfortunately, these molecules are mutation-dependent and are sometimes responsible for several side effects (such as hepatitis, abdominal pain) [8]. Even though pharmacology using CFTR potentiators is a real hope for most CF patients, rare mutations cannot currently be treated with those kinds of molecules.
2. Gene Therapy in CF Disease
Gene therapy may be a relevant approach to treat CF patients whatever their genotype. This technique was primarily used during the first trials of gene therapy in 1989–1990, which were coordinated by the NIH Clinical Center in Bethesda (Maryland) for adenosine deaminase (ADA) deficiency [9]. Since then, this technology kept on developing and became more and more efficient. The principle of gene transfer is based on the introduction of nucleic acids sequences. The plasmid, pDNA, containing a transgene encoding for the gene of interest, can be inserted into targeted cells through different systems. Viral vectors were the first to be used and have already proven efficient. In 2017, gene therapy was proved to be efficient in treating patients with hemophilia A and B using viral AAV vectors, providing a breakthrough to cure these monogenetic diseases [10][11]. The last gene therapy approved by the FDA and the EMA is Zolgensma (Novartis) for the treatment of spinal muscular atrophy [12]. However, viral vectors present some disadvantages. For instance, their immunogenicity limits the possibility of re-administration and they are difficult to produce and to purify. On the other hand, non-viral vectors are based on the use of chemical compounds, mostly lipids or cationic polymers, which directly cross the cytoplasmic membrane of the cells [13][14]. Compared to the viral strategy, the non-viral approach can easily be re-administrated thanks to the absence of an immunogenic reaction and to their good tolerance [15]. This re-administration is mandatory because of the constant renewal of epithelium pulmonary cells leading to the decrease in and eventually the disappearance of transgene expression. Moreover, non-viral vectors have the ability to compact longer nucleic acid sequences, which is useful for transgenes such as CFTR. However, the in vivo efficiency of non-viral vectors is still too low to get closer to the normal phenotype significantly, even if a change in the clinical state of the patient can be observed [15]. These strategies still have to be improved.
Given its characteristics, CF is a particularly complex and challenging model to approach pulmonary diseases and is a helpful model for other pathologies affecting the lungs and the respiratory tract. Successfully developing a gene therapy treatment for CF can potentially open the door to the treatment of other lung disorders. To directly target the lung, inhaled drugs can be administrated under different forms: pressurized metered-dose inhalers, nebulizers, soft mist inhalers, nasal sprays, and dry powders for inhalation (Figure 1A) [16][17]. The choice of devices has some consequences and influences the lung deposition [18]. This type of administration is a non-invasive method performed locally as drugs can easily reach the large absorptive surface area of the epithelium. Moreover, this technique has the benefit of avoiding the first-pass metabolism, intestinal efflux transporters, and degradation in the digestive system [19][20]. Deposition of an aerosol is dependent on the physical properties of generated micro-droplets. The droplet size determines the delivery location in pulmonary airways: the smaller the size, the deeper the deposition. The anatomy of the upper and lower airways and the breathing frequency are also key parameters that influence aerosol deposition [21][22]. Nebulization represents the process of transforming a liquid into an aerosol which can reach the respiratory tract (Figure 1B). This technique has the advantage of not requiring galenic formulation to a dry powder, which could damage the quality of the product. Qiu et al. showed that the lyophilization technique did not impact the in vitro transfection efficiency but, when tested on mice models using tracheal administration, nebulization of a reconstituted formulation was more efficient than the dry powder [23].
3. Current Challenges and Models
However, currently, over 25 clinical trials of gene delivery based on viral or non-viral gene vectors have failed to demonstrate significant clinical benefits, mainly due to inefficient gene transfer into the target cells [24]. Additionally, some viral CF gene therapy trials elicited host immune response. Consequently, clinical assays have been discontinuous, making subsequent treatments ineffective. Moreover, lifetime repeated treatment is required when using non-integrative systems for CF patients because of the transient nature of episomal transgene expression in addition to the natural life expectancy of the airways epithelial cells [25]. In 2015, the UK CF Gene Therapy Consortium completed a CF gene therapy clinical trial and observed that the use of a non-viral gene vector (GL67A, Genzyme corp.) had a significant, but still modest, benefit on the respiratory function (+3.7% of FEV1) compared to placebo controls [15]. They showed that a single aerosol per month, for one year, of a solution composed of nanoparticles (NPs) containing 13 mg of plasmid was very well tolerated. However, a more efficient gene delivery vector is needed to really make gene therapy a realistic option for the treatment of CF patients [15].
Extracellular barriers, indeed, have to be taken into account to adapt and finely tune their formulations (Figure 1B). Due to the presence of mucus as a protective barrier for xenobiotics, the passage through the cell is considerably reduced. The great majority of non-viral vectors as well as viral ones are stuck in the thick mucus of CF patients [26]. Inhaled gene vectors can be trapped within the mucus layer via non-covalent bonds (electrostatic and hydrophobic interactions) or simply cannot enter the network structure [26]. Many clinical studies have failed to demonstrate a gene therapy efficiency in the CF lung because the mucus was partially or totally neglected in in vivo and in vitro models. For instance, GL67A was selected using healthy sheep, and therefore without taking the mucus into account [27]. Moreover, animal models currently used for gene transfection studies are not totally satisfactory for CF model systems. For instance, CF mice and rats do not present spontaneous lung infections as well as mucus production [28]. W. K. O’Neal et al. successfully developed a transgenic mice model with overexpression of the ENaC β-subunit [29]. This model has the advantage of spontaneously exhibiting a CF-like lung disease more similar to human pathology with airways inflammation, bacterial infection, mucus dehydration, and obstruction [30][31]. Due to their anatomy, however, larger animal models represent better models to test inhaled gene therapy because of closer physiological and pathological similarities to human lungs. CFTR-knockout ferrets can be used as a potential model due to a lung structure close to that of humans. They exhibit altered chloride transport, leading to mucus hypersecretion and lung infections [32][33]. CF pigs also represent a potential model, as they present pulmonary manifestations similar to those found in human CF lung (infection, inflammation, airway remodeling, and mucus hypersecretion). They have, however, a short life expectancy (some days to few weeks) [34][35]. Nonetheless, every animal experimentation has to respect the 3R regulation (Reduce, Replace, and Refine). Difficulties in accessing animal models coupled with 3R restriction lead to the need for a more accurate cellular model. If the models used in vitro or in vivo are not sufficiently complex and representative of clinical conditions, the results obtained will not be reproducible during clinical trials. Therefore, it appears to be essential to have a reliable in vitro model close enough to the pathology studied to test and develop new vectorization strategies for nucleic acids with the ultimate aim of gene therapy. This review will focus on the use of mucus models and cellular cultures to obtain results closer to the in vivo pulmonary tracts and to avoid, as far as possible, failures in clinical development. Such models are fundamental to allow the development of non-viral gene carrier formulations able to overcome cellular and extracellular barriers faced in the respiratory tract.
References
- Kim, N.; Duncan, G.A.; Hanes, J.; Suk, J.S. Barriers to inhaled gene therapy of obstructive lung diseases: A review. J. Control. Release 2016, 240, 465, doi:10.1016/j.jconrel.2016.05.031.
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904, doi:10.1016/S0140-6736(09)60327-5.
- Ratjen, F.; Rietschel, E.; Griese, M.; Ballmann, M.; Kleinau, I.; Doring, G.; Reinhardt, D.; Paul, K. Fractional analysis of bron-choalveolar lavage fluid cytology in cystic fibrosis patients with normal lung function. Bronchoalveolar lavage for the evalu-ation of anti-inflammatory treatment (BEAT) study group. Eur. Respir. J. 2000, 15, 141–145.
- Reinhardt, N.; Chen, C.I.U.; Loppow, D.; Schink, T.; Kleinau, I.; Jörres, R.A.; Wahn, U.; Magnussen, H.; Paul, K.P. Cellular profiles of induced sputum in children with stable cystic fibrosis: Comparison with BAL. Eur. Respir. J. 2003, 22, 497–502, doi:10.1183/09031936.03.00043603.
- Jackson, A.D. Airway goblet-cell mucus secretion. Trends Pharmacol. Sci. 2001, 22, 39–45, doi:10.1016/S0165-6147(00)01600-X.
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531, doi:10.1016/S0140-6736(16)00576-6.
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819, doi:10.1056/NEJMoa1908639.
- Burgel, P.-R.; Munck, A.; Durieu, I.; Chiron, R.; Mely, L.; Prevotat, A.; Murris-Espin, M.; Porzio, M.; Abely, M.; Reix, P.; et al. Real-Life Safety and Effectiveness of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, doi:10.1164/rccm.201906-1227OC.
- Collins, F.S.; Gottlieb, S. The Next Phase of Human Gene-Therapy Oversight. N. Engl. J. Med. 2018, 379, 1393–1395, doi:10.1056/NEJMp1810628.
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530, doi:10.1056/NEJMoa1708483.
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227, doi:10.1056/NEJMoa1708538.
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Co-hort. J. Neuromuscul. Dis. 2019, 6, 307–317, doi:10.3233/JND-190403.
- Belmadi, N.; Midoux, P.; Loyer, P.; Passirani, C.; Pichon, C.; Le Gall, T.; Jaffres, P.-A.; Lehn, P.; Montier, T. Synthetic vectors for gene delivery: An overview of their evolution depending on routes of administration. Biotechnol. J. 2015, 10, 1370–1389, doi:10.1002/biot.201400841.
- Ia, P.; Sc, H.; Dr, G. Non-viral vectors in cystic fibrosis gene therapy: Recent developments and future prospects. Expert Opin. Biol. Ther. 2009, 9, 991–1003.
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Cal-cedo, R.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Repeated Nebulisation of Non-Viral Cystic Fibrosis Trans-membrane Conductance Regulator (CFTR) Gene Therapy in Patients with Cystic Fibrosis. Efficacy and Mechanism Evaluation; NIHR Journals Library: Southampton, UK, 2016.
- d’Angelo, I.; Conte, C.; La Rotonda, M.I.; Miro, A.; Quaglia, F.; Ungaro, F. Improving the efficacy of inhaled drugs in cystic fibrosis: Challenges and emerging drug delivery strategies. Adv. Drug Deliv. Rev. 2014, 75, 92–111, doi:10.1016/j.addr.2014.05.008.
- Ibrahim, M.; Verma, R.; Garcia-Contreras, L. Inhalation drug delivery devices: Technology update. Med. Devices (Auckland, NZ) 2015, 8, 131–139, doi:10.2147/MDER.S48888.
- Cheng, Y.S. Mechanisms of Pharmaceutical Aerosol Deposition in the Respiratory Tract. AAPS PharmSciTech 2014, 15, 630–640, doi:10.1208/s12249-014-0092-0.
- Audus, K.L.; Bartel, R.L.; Hidalgo, I.J.; Borchardt, R.T. The Use of Cultured Epithelial and Endothelial Cells for Drug Transport and Metabolism Studies. Pharm. Res. 1990, 7, 435–451, doi:10.1023/A:1015800312910.
- Ehrhardt, C.; Laue, M.; Kim, K.-J. In Vitro Models of the Alveolar Epithelial Barrier. In: Ehrhardt C., Kim KJ. (eds) Drug Ab-sorption Studies. Biotechnology: Pharmaceutical Aspects, vol VII. Springer, Boston, MA., USA, 2008; pp. 258–282, ISBN 978-0-387-74901-3.
- Byron, P.R.; Patton, J.S. Drug delivery via the respiratory tract. J. Aerosol Med. 1994, 7, 49–75, doi:10.1089/jam.1994.7.49.
- Resnier, P.; Mottais, A.; Sibiril, Y.; Le Gall, T.; Montier, T. Challenges and Successes Using Nanomedicines for Aerosol Deliv-ery to the Airways. Curr. Gene Ther. 2016, 16, 34–46.
- Qiu, Y.; Man, R.C.H.; Liao, Q.; Kung, K.L.K.; Chow, M.Y.T.; Lam, J.K.W. Effective mRNA pulmonary delivery by dry pow-der formulation of PEGylated synthetic KL4 peptide. J. Control. Release 2019, 314, 102–115, doi:10.1016/j.jconrel.2019.10.026.
- Griesenbach, U.; Alton, E.W.F.W. Gene transfer to the lung: Lessons learned from more than 2 decades of CF gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 128–139, doi:10.1016/j.addr.2008.09.010.
- Conese, M.; Boyd, A.C.; Di Gioia, S.; Auriche, C.; Ascenzioni, F. Genomic context vectors and artificial chromosomes for cystic fibrosis gene therapy. Curr. Gene Ther. 2007, 7, 175–187.
- Duncan, G.A.; Jung, J.; Hanes, J.; Suk, J.S. The Mucus Barrier to Inhaled Gene Therapy. Mol. Ther. 2016, 24, 2043–2053, doi:10.1038/mt.2016.182.
- Alton, E.W.F.W.; Baker, A.; Baker, E.; Boyd, A.C.; Cheng, S.H.; Coles, R.L.; Collie, D.D.S.; Davidson, H.; Davies, J.C.; Gill, D.R.; et al. The safety profile of a cationic lipid-mediated cystic fibrosis gene transfer agent following repeated monthly aero-sol administration to sheep. Biomaterials 2013, 34, 10267–10277, doi:10.1016/j.biomaterials.2013.09.023.
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for stud-ying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34, doi:10.1016/j.jcf.2017.09.001.
- Livraghi-Butrico, A.; Grubb, B.R.; Kelly, E.J.; Wilkinson, K.J.; Yang, H.; Geiser, M.; Randell, S.H.; Boucher, R.C.; O’Neal, W.K. Genetically determined heterogeneity of lung disease in a mouse model of airway mucus obstruction. Physiol. Genom. 2012, 44, 470–484, doi:10.1152/physiolgenomics.00185.2011.
- Livraghi-Butrico, A.; Kelly, E.J.; Klem, E.R.; Dang, H.; Wolfgang, M.C.; Boucher, R.C.; Randell, S.H.; O’Neal, W.K. Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial in-fection and inflammation. Mucosal Immunol. 2012, 5, 397–408, doi:10.1038/mi.2012.17.
- Livraghi-Butrico, A.; Kelly, E.J.; Wilkinson, K.J.; Rogers, T.D.; Gilmore, R.C.; Harkema, J.R.; Randell, S.H.; Boucher, R.C.; O’Neal, W.K.; Grubb, B.R. Loss of Cftr function exacerbates the phenotype of Na+ hyperabsorption in murine airways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L469–L480, doi:10.1152/ajplung.00150.2012.
- Fisher, J.T.; Liu, X.; Yan, Z.; Luo, M.; Zhang, Y.; Zhou, W.; Lee, B.J.; Song, Y.; Guo, C.; Wang, Y.; et al. Comparative Pro-cessing and Function of Human and Ferret Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 2012, 287, 21673–21685, doi:10.1074/jbc.M111.336537.
- Li, Z.; Engelhardt, J.F. Progress toward generating a ferret model of cystic fibrosis by somatic cell nuclear transfer. Reprod. Biol. Endocrinol. RBE 2003, 1, 83, doi:10.1186/1477-7827-1-83.
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR Gene Produces a Model of Cystic Fibrosis in Newborn Pigs. Science 2008, 321, 1837–1841, doi:10.1126/science.1163600.
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohl-ford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic Fibrosis Pigs Develop Lung Disease and Exhibit Defective Bacterial Eradication at Birth. Sci. Transl. Med. 2010, 2, 29ra31, doi:10.1126/scitranslmed.3000928.

