 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lijing Yang | + 2826 word(s) | 2826 | 2021-01-11 10:00:18 | | | |
| 2 | Vivi Li | Meta information modification | 2826 | 2021-01-13 03:45:44 | | | | |
| 3 | Vivi Li | Meta information modification | 2826 | 2021-02-07 09:58:55 | | |
Video Upload Options
Hematopoietic stem cells (HSCs) regularly produce various blood cells throughout life via their self-renewal, proliferation, and differentiation abilities. Most HSCs remain quiescent in the bone marrow (BM) and respond in a timely manner to either physiological or pathological cues, but the underlying mechanisms remain to be further elucidated. In the past few years, accumulating evidence has highlighted an intermediate role of inflammasome activation in hematopoietic maintenance, post-hematopoietic transplantation complications, and senescence. As a cytosolic protein complex, the inflammasome participates in immune responses by generating a caspase cascade and inducing cytokine secretion. This process is generally triggered by signals from purinergic receptors that integrate extracellular stimuli such as the metabolic factor ATP via P2 receptors. Furthermore, targeted modulation/inhibition of specific inflammasomes may help to maintain/restore adequate hematopoietic homeostasis.
1. Introduction
Hematopoiesis is a dynamic and continuous process involving the production of numerous immature and mature blood cells, which mostly relies on the self-renewal, proliferation, and differentiation abilities of HSCs [1]. The maintenance of HSCs is extracellularly regulated by the BM niche, which is mainly composed of hematopoietic cells, stromal cells, adipocytes, blood vessels, and nerves [2]. Determination and trafficking of HSCs can be stimulated by a variety of juxtacrine interactions (cell–cell or cell–matrix) or paracrine interactions (via cytokines, chemokines, or growth factors) associated with the HSC niche [2][3]. Most HSCs are quiescent in the BM under physiological conditions, and a small proportion of them occasionally divides to maintain self-renewal abilities and keep balance of the stem cell pool. Notably, under stress or pathological conditions such as hemorrhage or radiation exposure, HSCs are activated and enter the cell cycle in response to external challenges. However, how HSCs integrate external stimuli and respond appropriately requires further elaboration. New technologies, especially emerging single-cell analysis and cell fate tracing techniques, are continuously impacting the traditional understanding of the hierarchical model of hematopoiesis [4]. Multiple models of HSC development have been proposed to provide a greater understanding of hematopoiesis [5][6][7][8]. The consensus is that a complex network exists to orchestrate the hematopoietic process, meanwhile several proinflammatory signals have been demonstrated to be critical regulators of HSC development in the past few years. In this context, a deep understanding of how external cues such as infection, tissue damage, and physical stimuli impact HSC fate would be of great biological significance.
Ever since the term inflammasome was originally put forward in 2002, many studies have indicated that the inflammasome is an intracellular protein complex. The inflammasome is generally formed by a pattern recognition receptor (PRR), apoptosis-associated speck-like protein (ASC) and the inflammatory cysteine protease caspase-1 [9]. These supramolecular structures can be assembled in immune cell cytoplasm, resulting in systemic immune responses and inflammation. As requisite mediators of the innate immune response, inflammasomes serve as multiprotein scaffolds with two main functions: inflammatory reactions and systematic cell death [10][11][12]. Activation of inflammasomes promotes the maturation of the accumulating proinflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) through caspase-1 cleavage. IL-1β can also stimulate the release of other cytokines for example IL-1α, tumor necrosis factor (TNF)-α and IL-6 impacting the function of immune cells [13]. A cascade of downstream events originating from MyD88 recruitment by IL-1R or IL-18R will result in the activation of important signaling proteins and transcription factors, such as NF-κB, regulating inflammation [14]. Inflammasome activation also induces gasdermin D (GSDMD) cleavage by caspase-1, GSDMD as a key pyroptotic substrate of inflammatory caspases, the N-terminal of GSDMD fragment oligomerizes and inserts into the plasma membrane which induces pyroptosis [10][15][16][17]. Pyroptosis can be defined as a lytic form of programed cell death in response to external stimuli or host-derived danger signals, and it distinct from apoptosis by releasing inflammatory compounds into the extracellular space after cell swelling and membrane rupture [18].
Inflammation is a protective immune response that maintains homeostasis and involves various pathological processes, such as pathogen infection and tissue/organ damage. Several kinds of immune cells originating from HSCs constitute the foundation of the inflammatory response, and these cells are continuously replenished during infection to a certain extent [19]. Thus, understanding how HSCs respond to pathological alterations during inflammation is a meaningful research focus. Recent studies have also indicated that inflammasome activation during the inflammatory response plays an essential role in balancing multiple stages of hematopoietic homeostasis [20][21][22][23][24][25][26]. Both up- and down-regulation of inflammasome proteins can lead to a general inclination in homeostasis, suggesting that inflammasome activation may be required to carefully preserve hematopoiesis [21][27][28][29][30][31].
2. Relationship Between Inflammasomes and Hematopoiesis
The inflammasome assembles in response to danger signals, and inflammasome activation leads to inflammatory responses. There are two main types of signaling pathways involved in inflammasome activation: the canonical signaling pathway and the noncanonical signaling pathway. Numerous studies have suggested that the canonical signaling pathway, which was the first pathway discovered, plays a pivotal role in inflammatory responses and the pathogenesis of various inflammatory diseases [32][33]. There have only been a limited number of studies investigating the role of the noncanonical pathway in inflammatory responses, which mainly include murine caspase-11 activation and human caspase-4 and caspase-5 activation [34][35]. The canonical inflammasome pathway includes a group of nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) that is mainly composed of NLRP1, NLRP2, NLRP3, NLRP4, NLRP6, NLRP12, and absent in melanoma 2 (AIM2) [36]. Classically, canonical inflammasome activation is initiated by two kinds of signals and regulated at both the transcriptional and posttranslational levels. “Signal 1” is the priming signal and is associated with activation of the TLR/NF-κB pathway or mitochondrial-derived reactive oxygen species (ROS) that activate the TLR4/MyD88 signaling pathway. “Signal 2” can be induced by various stimuli, including pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), adenosine triphosphate (ATP), and uric acid crystals [29][37].
The hematopoietic system is classically divided into two major branches during the early stage of hematopoiesis: the myeloid lineage and lymphoid lineage; one prominent function of the myeloid lineage is to establish innate immunity [4][8][38]. Myeloid lineage cells in the circulation mainly include various innate immune cells, such as monocytes, neutrophils, eosinophils, basophils, and dendritic cells (DCs). These cells provide general defense against external pathogens and facilitate adaptive immune responses when they encounter various stimuli. Among lymphoid lineage cells, B cells and T cells participate in adaptive immunity [38][39]. Rapid adaptation of the hematopoietic stem/progenitor cell (HSPC) response to severe bacterial infection leads to peripheral blood (PB) neutrophilia and is defined as emergency granulopoiesis [20][23]. Such responses meet the increasing demand for the generation of immune cells, thus contributing to the chronicity of inflammatory diseases [40][41]. The significant role of inflammasomes in mediating the myeloid lineage and lymphoid lineage has been proven, especially in chronic metabolic diseases. Furthermore, the adaptation of HSPCs to inflammation has also been proven to be a critical event during the host response to infection, microenvironmental stress, or sterile inflammation [42].
2.1. Inflammasomes and HSPC Maintenance
BM has hematopoietic functions throughout life and mainly maintains a stable HSC pool. Accumulating evidence has suggested that the inflammasome is involved in different stages of hematopoiesis, and several kinds of inflammasome components have been demonstrated to impact HSPC maintenance [43][44].
Masters and colleagues first reported the pathophysiological effect of NLRP1 inflammasome activation on HSPCs [20]. The researchers noted that activation of the NLRP1α inflammasome in murine HSPCs induces a deadly systemic inflammatory disease that was driven by caspase-1 and IL-1β, independent of ASC and enhanced by IL-18 [20]. Activation of the NLRP1α inflammasome also triggers pyroptosis in HSPCs, resulting in leukopenia and BM hypoplasia, even in the absence of IL-1β-driven inflammation [20]. NLRP1α-deficient mice exhibit enhanced recovery from chemotherapy or viral infection, suggesting that the deletion of NLRP1α effectively increases the resistance of HSPCs to hematopoietic stresses [20]. This finding provides a potential intervention strategy for treating infection-induced cytopenias through which the competence of HSPCs under hematopoietic stress can be protected by removing or pharmacologically inhibiting NLRP1 inflammasome activation (Figure 1) [20]. Later, Hu et al. identified AIM2, another type of inflammasome that mediates HSPC death after whole-body irradiation in mice. The AIM2 inflammasome recruits ASC through its pyrin domain and forms an inflammasome to activate the canonical pathway in a cell-autonomous manner. AIM2-deficient mice are exempted from irradiation-induced hematopoietic failure, as AIM2 acts as a double-stranded DNA sensor that mediates the molecular mechanism of hematopoietic cell death in response to radiation-induced DNA damage (Figure 1). It has also been proposed that inhibiting AIM2 inflammasome activation (e.g., via MCC950, also known as CRID3) is a strategy to treat patients exposed to ionizing radiation due to events such as nuclear reactor leaks or radiotherapy [45].
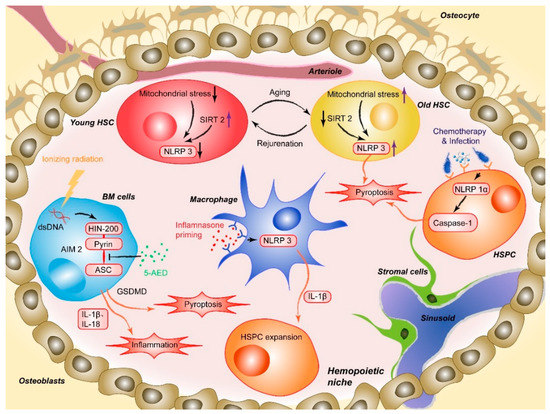
Figure 1. AIM2 is activated after the HIN200 domain senses cytoplasmic double-stranded DNA (dsDNA) caused by ionizing irradiation and the pyrin domain recruits the adapter protein ASC, which induces HSPC death. 5-AED inhibited radiation-induced AIM2 inflammasome activation by decreasing the interaction between AIM2 and ASC. Mitochondrial stress initiates aberrant activation of the NLRP3, and SIRT2 activation inhibits the activation of the NLRP3 inflammasome in HSCs, which suggests a method for reversing aging HSCs. NLRP1α inflammasome activation through chemotherapy or viral infection induces HSPC pyroptosis, resulting in cytopenia, including leukopenia and BM hypoplasia, in the absence of IL-1β-driven inflammation. NLRP3 inflammasome-mediated IL-1β signaling in macrophages drives HSPC production in response to metabolic activity.
Moreover, activation of some lineage-specific transcription factors is imperative for mediating the response of HSPCs to infection or sterile inflammation [46]. Metabolic activity has been described as a critical factor regulating stem cell fate decisions, proliferation, and differentiation. In a recently published paper, Frame et al. demonstrated that NLRP3 inflammasome-mediated IL-1β signaling within macrophages in response to metabolic alterations could be an enhancing factor to drive HSPC production in a zebrafish model (Figure 1). The inflammasome serves as a metabolic sensor to trigger IL-1β production and expand developing HSPCs, while up- or down-regulation of the NLRP3 inflammasome accordantly changes the production of HSPCs inside zebrafish embryos or human HSPC cultures [24]. Changes in external conditions can also impact the adaptation of HSPCs. For example, BM cells and hematopoiesis can be severely affected by high-dose radiotherapy [47][48][49]. The process involves a series of cellular and molecular changes, such as the depletion of hematopoietic cells, proinflammatory cytokine and chemokine release, activation and destruction of peripheral immune cells, and DNA damage [49][50]. Accumulating evidence has shown that inflammasome activation plays an important role in mediating radiation-induced cell and tissue damage [51][52]. Specifically, various recruited cells during radiation-induced damage, especially macrophages, are activated to undergo pyroptosis through the NLRP3-caspase-1 axis. Knockout of NLRP3 protected mice from radiation-induced macrophage pyroptosis by suppressing caspase-1 activation [51]. Moreover, given the abovementioned radiation-induced upregulation of the AIM2 inflammasome, the latest research by Wu et al. suggested that 5-androstenediol (5-androstene-3β-17β-diol, 5-AED), a natural steroid hormone produced by the adrenal cortex, could markedly attenuate irradiation-induced AIM2 inflammasome activation, promoting the survival of mice. Subcutaneous administration of 5-AED enhances the recovery of the hematopoietic system and decreases tissue damage by promoting NF-κB signaling and inhibiting inflammasome-mediated pyroptosis possibly by disrupting the interaction between AIM2 and ASC (Figure 1) [53]. A study by Li et al. illustrated that 2-Gy irradiation increased the protein expression levels of NLRP3 in THP-1 cells and elevated ROS levels [54].
While these studies documented the role of inflammasome activation in inhibiting hematopoiesis, activation of another inflammasome subtype also seems to have some positive effects on hematopoiesis. Linz et al. demonstrated that NLRP12 profoundly impacts hematopoietic recovery by suppressing TNF signaling in vivo during emergency hematopoiesis induced by the combination of radiation exposure and thermal injury. As a checkpoint of TNF signaling, the NLRP12 inflammasome functionally limits TNF-induced HSPC apoptosis, and it has been proven that inflammation in the absence of NLRP12 participation leads to HSPC apoptosis, as well as defective peripheral immune reconstitution. In addition, myelopoiesis and immune cell reconstitution are also accelerated by NLRP12 overexpression [23]. Du et al. illustrated that chronic DNA damage upregulates the NLRP12 inflammasome in HSPCs from Fanca−/− mice. In a newly published paper, the researchers further investigated the essential role of NLRP12 in HSC maintenance and found that persistent DNA damage-induced NLRP12 improves HSC function in both mouse and human models of DNA repair deficiency (Fanca−/− mice). Functionally, knockdown of NLRP12 exacerbates the repopulation defect in Fanca−/− HSCs, and overexpression of NLRP12 substantially improves the long-term repopulating function of Fanca−/− HSCs, suggesting a potential genetic or pharmacological strategy to target the NLRP12 inflammasome to obtain therapeutic effects [22][55]. In fact, the lineage contribution of HSPCs in hematopoiesis is no less than that of HSCs, and HSPCs serve as active players in the innate immune response to systemic stimuli, including DNA damage. We hypothesize that HSPCs exert a similar function in Fanca−/− mice as HSCs.
Collectively, the abovementioned effects of different inflammasomes on the maintenance of hematopoiesis still need to be further addressed regardless of physiology or pathology. Consistent with the existing research, we believe there is an interactive transcription network through which signals converge and subsequently regulate inflammasome activity to maintain steady-state hematopoiesis [56][57][58][59][60][61].
2.2. Inflammasomes and HSPC Differentiation
Differentiation refers to the process by which progenitor cells develop the appearance of mature PB cells, and the construction of a hierarchical system has gained much attention. Initial lineage priming of the differentiation process is strictly managed by gene-expression modules regulated by lineage-specific transcription factors [62][63]. Some studies have shown that the inflammasome and its components also play a decisive role in HSPC differentiation [57][58][64].
Earlier studies have proven that caspase activation is closely related to the differentiation of several myeloid lineages; as a critical transcription factor, GATA-1 controls erythroid differentiation and pro-platelet formation and maturation [65]. GATA-1 is preferentially localized in the nucleus through an elaborate balance achieved by the interaction between caspase-3 and chaperone HSP70 to prevent cleavage [66]. Under conditions in there is an acute need for platelets, caspase-3 can be activated in response to IL-1α, thus promoting the formation of platelets. IL-1β is usually secreted by monocytes in response to lipopolysaccharide. through an inflammasome activation-dependent pathway. A recent report from the Tyrkalska group supported this view, indicating that inflammasomes participate in erythroid/myeloid cell fate decisions and that terminal erythroid differentiation in chronic inflammatory diseases eventually contributes to hematopoietic bias [21][66]. Pharmacological inhibition of the inflammasome ameliorated neutrophilic inflammation and anemia in zebrafish disease models. GATA-1 is increased in inflammasome-deficient larvae and is responsible for facilitating erythropoiesis and inhibiting myelopoiesis. Interestingly, inflammasome inhibition did not affect the granulocyte-monocyte myeloid transcription factor PU.1 (SPI1) level, indicating that there are some indirect effects left to explore. These results show that the inflammasome plays an essential role in the pathogenesis of neutrophilia and anemia during chronic inflammatory diseases, suggesting a pharmacological target for therapeutic interventions [21].
Acute myeloid leukemia (AML) is characterized by the blockade of hematopoietic differentiation and cell death, and interesting work from the Jost laboratory demonstrated that receptor-interacting protein kinase 3 (RIPK3) promotes the differentiation of leukemia-initiating cells by activating the inflammasome. RIPK3 suppresses malignant myeloproliferation by activating the inflammasome, thus promoting differentiation and cell death, and RIPK3 expression is often reduced in primary de novo AML to prevent leukemia-initiating cells from dying [67].
2.3. Inflammasomes and Aging-Associated Hematopoiesis
Aging is an unavoidable consequence of life, and enhanced myelopoiesis is a hallmark of BM aging and impaired lymphopoiesis, which are mainly caused by myeloid-biased HSPC proliferation and differentiation. This alteration in hematopoiesis is sometimes referred to in the literature as inflamm-aging, which is the chronic, low-grade sterile inflammation that is present in advanced age and manifests some relevant clinical symptoms.
The Dixit group first demonstrated the role of the NLRP3 inflammasome in promoting age-related thymic atrophy and immune senescence. The researchers found that deletion of the inflammasome components NLRP3 and ASC significantly increased the number of cortical thymic epithelial cells and T cell progenitors, which reduced aging-related thymic atrophy. The deletion also accelerated T cell reconstitution and immune recovery in middle-aged animals, suggesting an NLRP3 inflammasome-dependent mechanism through thymic caspase-1 activation mediates this process [68][69]. Recently, another study showed that the NLRP3 inflammasome was aberrantly activated in HSCs during physiological aging. This activation was mainly mediated by mitochondrial stress and SIRT2 inactivation, contributing to the functional decline in aging HSCs. As a cytosolic NAD-dependent deacetylase, SIRT2 is required for HSC maintenance and regenerative capacity during senescence by suppressing the activation of the NLRP3 inflammasome in HSCs [69][70][71].+
Luo et al. have demonstrated that SIRT2 regulates the functional deterioration of HSCs in aging models by repressing the NLRP3 inflammasome activation, which SIRT2 activation, NLRP3 inflammasome inactivation or caspase-1 inactivation improves the maintenance and regenerative capacity of aged HSCs [72]. Functionally, overexpression of SIRT2 can increase the maintenance and regenerative capacity of aged HSCs, which did not significantly influence young HSCs. Thus, these results indicate a potential SIRT2-NLRP3-caspase-1 axis in which the function of senescent hematopoietic and immune cells can be maintained or even rejuvenated. In contrast, in the previously described study, aging-related, persistent DNA damage-induced NLRP12 expression improved HSC function in both mouse and human models of DNA repair deficiency (Fanca−/− mice). The authors found that the depletion of NLRP12 in aged HSCs compromised their self-renewal and hematopoietic recovery capacities, suggesting that pharmacological activation of NLRP12 may have therapeutic value in enhancing the function of aged HSCs [22][55].
References
- Qiu, J.; Papatsenko, D.; Niu, X.; Schaniel, C.; Moore, K. Divisional history and hematopoietic stem cell function during homeostasis. Stem. Cell Rep. 2014, 2, 473–490.
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648.
- Gomes, C.A.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231.
- Carrelha, J.; Meng, Y.; Kettyle, L.M.; Luis, T.C.; Norfo, R.; Alcolea, V.; Boukarabila, H.; Grasso, F.; Gambardella, A.; Grover, A.; et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 2018, 554, 106–111.
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.J.; Thoren, L.A.; et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306.
- Perie, L.; Duffy, K.R.; Kok, L.; de Boer, R.J.; Schumacher, T.N. The Branching Point in Erythro-Myeloid Differentiation. Cell 2015, 163, 1655–1662.
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116.
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216.
- de Rivero Vaccari, J.P. The Inflammasome in Reproductive Biology: A Promising Target for Novel Therapies. Front. Endocrinol. 2020, 11, 8.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298.
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287.
- Dinarello, C.A. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 2011, 41, 1203–1217.
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012, 119, 2991–3002.
- Masters, S.L.; Gerlic, M.; Metcalf, D.; Preston, S.; Pellegrini, M.; O’Donnell, J.A.; McArthur, K.; Baldwin, T.M.; Chevrier, S.; Nowell, C.J.; et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 2012, 37, 1009–1023.
- Tyrkalska, S.D.; Perez-Oliva, A.B.; Rodriguez-Ruiz, L.; Martinez-Morcillo, F.J.; Alcaraz-Perez, F.; Martinez-Navarro, F.J.; Lachaud, C.; Ahmed, N.; Schroeder, T.; Pardo-Sanchez, I.; et al. Inflammasome Regulates Hematopoiesis through Cleavage of the Master Erythroid Transcription Factor GATA1. Immunity 2019, 51, 50–63.e5.
- Lin, Q.; Wu, L.; Ma, Z.; Chowdhury, F.A.; Mazumder, H.H.; Du, W. Persistent DNA damage-induced NLRP12 improves hematopoietic stem cell function. JCI Insight 2020, 5.
- Linz, B.M.; Neely, C.J.; Kartchner, L.B.; Mendoza, A.E.; Khoury, A.L.; Truax, A.; Sempowski, G.; Eitas, T.; Brickey, J.; Ting, J.P.; et al. Innate Immune Cell Recovery Is Positively Regulated by NLRP12 during Emergency Hematopoiesis. J. Immunol. 2017, 198, 2426–2433.
- Frame, J.M.; Kubaczka, C.; Long, T.L.; Esain, V.; Soto, R.A.; Hachimi, M.; Jing, R.; Shwartz, A.; Goessling, W.; Daley, G.Q.; et al. Metabolic Regulation of Inflammasome Activity Controls Embryonic Hematopoietic Stem and Progenitor Cell Production. Dev. Cell 2020, 55, 133–149.e136.
- Ratajczak, M.Z.; Adamiak, M.; Thapa, A.; Bujko, K.; Brzezniakiewicz-Janus, K.; Lenkiewicz, A.M. NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow. Leukemia 2019, 33, 815–825.
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214.
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181.
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975.
- Ratajczak, M.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.; Kucia, M. The Nlrp3 inflammasome as a "rising star" in studies of normal and malignant hematopoiesis. Leukemia 2020, 34, 1512–1523.
- Stoilova, B.; Vyas, P. The Inflammasome: More Than a Protective Innate Immune Mechanism. Immunity 2019, 51, 3–5.
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847.
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36.
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192.
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121.
- Monie, T.P. The Canonical Inflammasome: A Macromolecular Complex Driving Inflammation. Subcell. Biochem. 2017, 83, 43–73.
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622.
- Liggett, L.A.; Sankaran, V.G. Unraveling Hematopoiesis through the Lens of Genomics. Cell 2020, 182, 1384–1400.
- Pucella, J.N.; Upadhaya, S.; Reizis, B. The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu. Rev. Cell Dev. Biol. 2020, 36, 529–550.
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314.
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720.
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019, 20, 802–811.
- Orelio, C.; Haak, E.; Peeters, M.; Dzierzak, E. Interleukin-1-mediated hematopoietic cell regulation in the aorta-gonad-mesonephros region of the mouse embryo. Blood 2008, 112, 4895–4904.
- Orelio, C.; Peeters, M.; Haak, E.; van der Horn, K.; Dzierzak, E. Interleukin-1 regulates hematopoietic progenitor and stem cells in the midgestation mouse fetal liver. Haematologica 2009, 94, 462–469.
- Hu, B.; Jin, C.C.; Li, H.B.; Tong, J.Y.; Ouyang, X.S.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768.
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618.
- Xiao, J.; Wang, C.; Yao, J.C.; Alippe, Y.; Yang, T.; Kress, D.; Sun, K.; Kostecki, K.L.; Monahan, J.B.; Veis, D.J.; et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020, 18, e3000807.
- Huang, S.; Che, J.; Chu, Q.; Zhang, P. The Role of NLRP3 Inflammasome in Radiation-Induced Cardiovascular Injury. Front Cell Dev. Biol. 2020, 8, 140.
- Buckley, A.M.; Lynam-Lennon, N.; O’Neill, H.; O’Sullivan, J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 298–313.
- Huang, Y.; Zhang, W.; Yu, F.; Gao, F. The Cellular and Molecular Mechanism of Radiation-Induced Lung Injury. Med. Sci. Monit. 2017, 23, 3446–3450.
- Liu, Y.G.; Chen, J.K.; Zhang, Z.T.; Ma, X.J.; Chen, Y.C.; Du, X.M.; Liu, H.; Zong, Y.; Lu, G.C. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 2017, 8, e2579.
- Wei, J.; Wang, H.; Wang, H.; Wang, B.; Meng, L.; Xin, Y.; Jiang, X. The role of NLRP3 inflammasome activation in radiation damage. Biomed. pharmacother. 2019, 118, 109217.
- Wu, T.; Liu, W.; Fan, T.; Zhong, H.; Zhou, H.; Guo, W.; Zhu, X. 5-Androstenediol prevents radiation injury in mice by promoting NF-kappaB signaling and inhibiting AIM2 inflammasome activation. Biomed. Pharm. 2020, 121, 109597.
- Li, X.Y.; Gong, Y.L.; Li, D.; Xiang, L.; Ou, Y.H.; Jiang, L.; Shu, P.; Liu, X.K.; Guo, F.C.; Qin, D.Y.; et al. Low-Dose Radiation Therapy Promotes Radiation Pneumonitis by Activating NLRP3 Inflammasome. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 804–814.
- Du, W.; Amarachintha, S.; Wilson, A.; Pang, Q. The immune receptor Trem1 cooperates with diminished DNA damage response to induce preleukemic stem cell expansion. Leukemia 2017, 31, 423–433.
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62.
- Espin-Palazon, R.; Stachura, D.L.; Campbell, C.A.; Garcia-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 2014, 159, 1070–1085.
- Espin-Palazon, R.; Weijts, B.; Mulero, V.; Traver, D. Proinflammatory Signals as Fuel for the Fire of Hematopoietic Stem Cell Emergence. Trends Cell Biol. 2018, 28, 58–66.
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, D.; Lv, J.; Liu, F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 2015, 125, 1098–1106.
- Harris, J.M.; Esain, V.; Frechette, G.M.; Harris, L.J.; Cox, A.G.; Cortes, M.; Garnaas, M.K.; Carroll, K.J.; Cutting, C.C.; Khan, T.; et al. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood 2013, 121, 2483–2493.
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Giladi, A.; Paul, F.; Herzog, Y.; Lubling, Y.; Weiner, A.; Yofe, I.; Jaitin, D.; Cabezas-Wallscheid, N.; Dress, R.; Ginhoux, F.; et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 2018, 20, 836–846.
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281.
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800.
- Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. Down-regulation of GATA1 uncouples STAT5-induced erythroid differentiation from stem/progenitor cell proliferation. Blood 2010, 115, 4367–4376.
- De Maria, R.; Zeuner, A.; Eramo, A.; Domenichelli, C.; Bonci, D.; Grignani, F.; Srinivasula, S.M.; Alnemri, E.S.; Testa, U.; Peschle, C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 1999, 401, 489–493.
- Hockendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91.
- Youm, Y.H.; Kanneganti, T.D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012, 1, 56–68.
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591.
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286.
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. SIRT7 Represses Myc Activity to Suppress ER Stress and Prevent Fatty Liver Disease. Cell Rep. 2013, 5, 654–665.
- Luo, H.; Mu, W.C.; Karki, R.; Chiang, H.H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954.e944.


